КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Лекция 15. Слабые и сильные электролиты
|
|
|
|
1. Электролитическая ионизация. Степень ионизации. Константа ионизации.
2. Понятие о теории сильных электролитов. Активность.
3. Кислотно-основная ионизация.
1. Электролитическая ионизация. Степень ионизации. Константа ионизации
Изучение разбавленных растворов показало, что все их общие свойства (понижение давления пара, изменение температур замерзания и кипения, величина осмотического давления) изменяются пропорционально числу частиц растворенного вещества. Эта формулировка представляет собой обобщенный закон разбавленных растворов Рауля–Вант-Гоффа. Эта общая закономерность оказалась справедливой для растворов органических веществ в воде и для растворов в органических растворителях. При исследовании водных растворов солей, кислот, оснований было обнаружено, что изменение соответствующего свойства в зависимости от состава раствора значительно превышает ожидаемую величину. Например, понижение температуры замерзания моляльного раствора NaCl превышает почти в два раза криоскопическую постоянную для воды (3,36° вместо 1,86°). Это свидетельствует о том, что число частиц в водных растворах кислот, оснований и солей не соответствует молярной концентрации раствора.
Кроме того, растворы, для которых характерны отклонения от законов разбавленных растворов, обладают значительной электропроводностью в отличие от водных растворов некоторых органических веществ. Это можно было объяснить наличием в растворе заряженных частиц. Растворы, проводящие электрический ток, были названы электролитами.
Свойства электролитов были рассмотрены и обобщены основоположником теории электролитической ионизации Аррениусом (1887) и развиты в трудах В. А. Кистяковского, И. А. Каблукова на основе химической (гидратной) теории растворения Д. И. Менделеева. Основные положения теории электролитической ионизации:
1) при растворении солей, кислот и оснований в воде происходит диссоциация этих веществ с образованием электрически заряженных частиц – катионов и анионов;
2) электропроводность таких растворов прямо пропорциональна общей концентрации ионов в растворе.
В работах И. А. Каблукова и В. А. Кистяковского отмечалось, что электролитическая ионизация вызывается взаимодействием полярных молекул растворителя с частицами растворяемого вещества (молекулами газов, атом-ионами при растворении кристаллов). Это взаимодействие приводит к поляризации даже преимущественно ковалентных связей, как, например, в хлористом водороде. При растворении этого газа в воде происходит образование ионов водорода и хлора за счет ослабления связи Н—С1 в среде с большой диэлектрической постоянной. Переход ионов в раствор сопровождается их гидратацией:
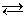 НС1 + nН2О H+ (Н2О) x + С1- (Н2О) n - x
НС1 + nН2О H+ (Н2О) x + С1- (Н2О) n - x
Такой же процесс наблюдается и при растворении ионно-ковалентных кристаллов (например, NaCI) в воде. Хотя в кристаллической решетке NaCl нет ионов Na+ и Сl-, однако взаимодействие с дипольными молекулами растворителя способствует поляризации связей в кристалле, их ослаблению и обеспечивает возможность перехода частиц в раствор с образованием гидратированных ионов:
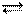 НС1 + nН2О Na+ (Н2О) x + С1- (Н2О) n - x
НС1 + nН2О Na+ (Н2О) x + С1- (Н2О) n - x
Таким образом, в сильно полярных растворителях ионизируются не только вещества с преимущественно гетерополярной связью (соли), но и молекулы, характеризующиеся малой ионностью. С этой точки зрения известное правило “подобное растворяется в подобном” не является универсальным.
Процесс гидратации сильно экзотермичен и идет самопроизвольно с уменьшением энтальпии. Теплота гидратации заметно превышает энергию разрыва связи (теплоту диссоциации). Обычно степень гидратации, т. е. количество молекул растворителя, окружающих каждый ион, очень велико (n и х – целые числа, n >> 1, x >>1); лишь при ионизации кислоты х = 1:
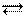 НA + nН2О H3O+ + A-(Н2О) n -1
НA + nН2О H3O+ + A-(Н2О) n -1
Это объясняется малым размером иона водорода (протона), который составляет ~10-4 от размера атома. При гидратации протон внедряется в сферу молекулы H2O с образованием оксоний-иона Н3О+. Новая ковалентная связь образуется по донорно-акцепторному механизму за счет свободной электронной пары кислорода и является насыщенной. Одной из количественных характеристик электролитической ионизации является степень ионизации, которая определяется как отношение диссоциированных молекул к общему числу растворенных молекул. Обычно степень ионизации выражают в долях единицы или в процентах:
α = (n / n 0)100,
где n 0 – число растворенных частиц; n – число частиц, подвергшихся электролитической ионизации.
По степени ионизации электролиты условно подразделяются на сильные (α > 30%) и слабые (α < 3%). Степень ионизации зависит от природы растворителя. Чем более полярна молекула растворителя, тем при прочих равных условиях выше степень ионизации растворенного вещества. Поскольку электролитическая ионизация сопровождается тепловым эффектом, то степень ионизации зависит от температуры, причем влияние температуры можно оценить по принципу Ле-Шателье: если электролитическая ионизация представляет собой эндотермический процесс, то с повышением температуры степень ионизации растет, в противоположном случае — уменьшается.
Сильно влияет на степень электролитической ионизации концентрация раствора. Если рассматривать электролитическую ионизацию как равновесный обратимый химический процесс
 KA + nН2О K+ (Н2О) x + A- (Н2О) n - x,
KA + nН2О K+ (Н2О) x + A- (Н2О) n - x,
то в соответствии с принципом смещения равновесия разбавление водой увеличивает количество диссоциированных молекул, т. е. степень ионизации при разбавлении возрастает. В связи с этим деление электролитов по силе в соответствии с величиной степени ионизации условно и приведенная классификация справедлива только для 0,1 н. растворов.
Процесс электролитической ионизации удобнее характеризовать константой ионизации, применив к нему законы химического равновесия. Так, для реакции
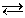 КА К+ + А-
КА К+ + А-
константа ионизации Ки = [К+][А-]/[КА]. Здесь и далее символом [ ] обозначаются молярные концентрации компонентов. В отличие от степени ионизации константа электролитической ионизации зависит лишь от природы электролита и температуры. Чем больше величина Ки, тем сильнее электролит.
Между константой и степенью электролитической ионизации существует количественная связь. Действительно, пусть в рассмотренном процессе общее количество растворенного вещества КА равно С, а степень ионизации равна α. Тогда [К+] = [А-] = αС и, соответственно, концентрация недиссоциированных частиц [КА] = (1 – α) С. Подставив эти значения в выражение для константы ионизации, получим:
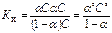
Полученное соотношение известно под названием закона разбавления Оствальда.
Для слабых электролитов, когда α << 1, Ки» α2 С. Отсюда
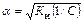
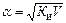
где V – разбавление,
Из формулы следует, что если, например, разбавить раствор в 100 раз, то степень ионизации возрастет в 10 раз.
С учетом степени электролитической ионизации можно применить законы разбавленных растворов и к растворам электролитов введением поправочного множителя i, называемого изотоническим коэффициентом Вант-Гоффа. Тогда отношение соответствующего свойства (понижение давления пара, изменение температуры плавления и кипения, осмотическое давление) для электролита к аналогичному свойству раствора неэлектролита той же концентрации равно коэффициенту Вант-Гоффа, т.е.
∆P’/∆P = ∆T’3/∆T3 = ∆T’k/∆Tk = P’осм/Росм = i
Очевидно, что для растворов электролитов всегда i > 1, а для растворов неэлектролитов i = 1. Законы разбавленных растворов могут быть легко трансформированы применительно к растворам электролитов:
P’осм. = iСRT, DTзам. = i×K×Сm, DTкип. = i×E×Сm, P’ = iP0AxA
Изотонический коэффициент можно связать со степенью ионизации раствора электролита. Пусть степень ионизации некоторого электролита с общим числом молекул в растворе С равна α. Предположим, что при диссоциации каждая молекула электролита распадается на n ионов. Тогда число молекул электролита, распавшихся на ионы, равно α С, число ионов в растворе – n α С, а число молекул, не распавшихся на ионы, – (1 – α) С. Общее число частиц в растворе равно (1 – α) С + n α С. Отношение общего числа частиц в растворе к числу растворенных молекул представляет собой изотонический коэффициент:
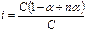 , i=1 – α (1– n).
, i=1 – α (1– n).
α = (i – 1)/(n – 1).
Данное соотношение позволяет определить степень ионизации электролита по отклонению его свойств от законов разбавленных растворов. В качестве примера найдем степень ионизации 0,1 н. K2SO4, который замерзает при -0,225° С (DТЗ' = 0,225°). Для водных растворов неэлектролитов криоскопическая константа равна К = 1,86°. Если бы K2SO4 не распадался на ионы в водном растворе, то соответствующее понижение 0,1 н. раствора было бы равно 0,093°. Тогда изотонический коэффициент 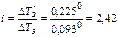 . Поскольку каждая молекула K2SO4 распадается на 3 иона: K2SO4 → 2K+ + SO42-(n = 3), то
. Поскольку каждая молекула K2SO4 распадается на 3 иона: K2SO4 → 2K+ + SO42-(n = 3), то
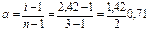 (0,71%)
(0,71%)
т.е. K2SO4 в 0,1 н. растворе диссоциирован примерно на 71%.
2. Понятие о теории сильных электролитов. Активность
Для сильных электролитов, когда степень ионизации велика, константа ионизации зависит от концентрации, так как при накоплении в растворе большого числа ионов сказывается их взаимное влияние.
Потенциально свойствами сильных электролитов обладают вещества, имеющие кристаллическую структуру координационного типа со значительной ионностью связи. Типичным примером подобных веществ являются многие соли. В их кристаллической решетке невозможно выделить отдельную молекулу. Поэтому при растворении таких веществ в полярных растворителях (вода) в раствор переходят отдельные сольватированные ионы, и, таким образом, процесс электролитической ионизации протекает полностью, т.е. недиссоциированные частицы в растворе отсутствуют. Отсюда следует, что для растворов сильных электролитов неприменимы представления о константе и степени ионизации, так как оба эти понятия учитывают присутствие в растворе некоторой доли недиссоциированных частиц.
При определенных условиях, например когда растворитель обладает малой диэлектрической проницаемостью, создаются условия для электростатического взаимодействия сольватированных ионов противоположного знака. При этом последние подходят друг к другу на близкое расстояние и образуют так называемую ионную пару – сложный агрегат, состоящий из двух противоположно заряженных ионов, окруженных молекулами растворителя, в котором электрические заряды взаимно компенсированы. Такой процесс называется ассоциацией. По своей природе и механизму образования ионные пары не тождественны недиссоциированным молекулам слабых электролитов.
Представление об образовании ионных пар в растворах сильных электролитов было введено Бьёррумом и Семенченко. В соответствии с этой концепцией для каждого растворителя существует определенный параметр q (параметр Бьёррума), представляющий собой расстояние, на которое подходят друг к другу ионы в процессе образования ионной пары. Этот параметр определяется из соотношения:
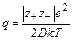
где z+, z_ – заряды катиона и аниона;
е – заряд электрона = 4,8∙10-10 эл.ст.ед. (см3/2∙г1/2 ∙ с-1);
k – константа Больцмана = 1,38∙10-16 эрг/град (г∙см2∙с-2 ∙град-1);
T – абсолютная температура,
К; D – диэлектрическая проницаемость растворителя.
Из соотношения следует, что при увеличении заряда ионов расстояние, на котором они начинают взаимодействовать, увеличивается. Наоборот, при увеличении диэлектрической проницаемости растворителя сила электростатического взаимодействия между ионами уменьшается в D раз. Поэтому полярные растворители, характеризующиеся большим значением диэлектрической проницаемости, способствуют образованию растворов, являющихся сильными электролитами с малой склонностью к образованию ионных пар. Даже на сравнительно малых расстояниях взаимодействием ионов можно пренебречь (q мало по величине), в силу чего ионы можно считать практически изолированными. При увеличении температуры параметр Бьёррума q уменьшается и взаимодействие между ионами ослабляется на меньших расстояниях, что объясняется возрастанием энергии теплового движения ионов. Параметр Бьёррума имеет вполне определенное значение для каждого растворителя при заданных температуре и заряде ионов. Например, для однозарядных ионов в воде (z+ = z - =1) при температуре 25° С = 298 К
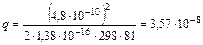 см = 3,57 Å
см = 3,57 Å
Если расстояние между ионами меньше этой величины, то молекулу можно считать недиссоциированной. Если же q > 3,57 Å, ионы рассматриваются как изолированные.
В разбавленных растворах сильных электролитов ионы находятся на расстояниях, значительно превышающих параметр Бьёррума, между собой не взаимодействуют и при этом электролит ионизирован полностью. При повышении концентрации раствора расстояния между ионами сокращаются, что усиливает межионное взаимодействие. Вследствие этого экспериментально определяемые свойства растворов сильных электролитов (D Р, D T кип, D Т з и т.п.), зависящие от общего количества частиц в растворе, оказываются меньше рассчитанных в предположении полной ионизации. Так, при ионизации K2SO4 теоретическое значение изотонического коэффициента должно быть равно 3 (поскольку каждая формульная единица K2SO4 распадается в растворе на 3 иона). Экспериментальная величина изотонического коэффициента, определенная по понижению температуры замерзания раствора, равна 2,42. Вследствие этого кажущаяся степень ионизации α = 71%. Создается впечатление, что ионизация прошла не полностью и в растворе имеется некоторое количество недиссоциированных частиц. На самом деле этот эффект обусловлен ассоциацией сольватированных ионов с образованием ионных пар. Именно поэтому степень ионизации в растворах сильных электролитов, определяемая экспериментально, является кажущейся. Таким образом, для растворов сильных электролитов законы идеальных растворов оказываются неприменимыми. Количественное описание поведения таких растворов осложняется многими факторами, определяющими общее число частиц в растворе.
Чтобы можно было пользоваться простыми соотношениями идеальных растворов для описания поведения реальных растворов, Льюис в 1907 г. ввел формальное представление об эффективной концентрации – активности. Активность связана с истинной концентрацией растворенного вещества соотношением
α = g С,
где α – активность; С – концентрация; g – коэффициент активности.
Активность измеряется в тех же единицах, что и концентрация, поскольку коэффициент активности – величина безразмерная. Он характеризует степень отклонения свойств данного раствора от свойств идеального раствора. Для бесконечно разбавленных растворов электролитов, где практически отсутствует взаимодействие ионов, активность становится равной концентрации и коэффициент активности равен единице:
g = α/ С = 1.
Если вместо концентрации в уравнения, отражающие законы Рауля, Генри, Вант-Гоффа и др., подставить экспериментально определённые значения активности, то эти уравнения остаются справедливыми и для реальных растворов, в частности для растворов сильных электролитов.
Введение понятия об активности позволяет, не выясняя сложной картины взаимодействия частиц в реальном растворе, оценить суммарный эффект этого взаимодействия, проявляющийся в отклонении свойств системы от идеальной, и применять законы идеальных растворов для анализа реальных систем.
3. Кислотно-основная ионизация
Характер электролитической ионизации гидроокисей общей формулы ЭОН зависит от сравнительной прочности и полярности связей Э—О и О—Н и может протекать по двум типам:
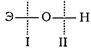
 ЭОН Э+ + ОН- (I)
ЭОН Э+ + ОН- (I)
 ЭOH ЭO- + H+ (II)
ЭOH ЭO- + H+ (II)
Полярность связей, как известно, определяется разностью элект-роотрицательностей компонентов, размерами и эффективным зарядом атомов. Щелочные и щелочноземельные металлы, а также переходныеэлементы в низших степенях окисления образуют ионы относительно большого размера и с малым эффективным зарядом. Поэтому удельный заряд таких ионов невелик и их поляризующие свойства выражены слабо. При этом связь Э—О обладает сравнительно малой прочностью и диссоциация ЭОН идет преимущественно за счет отщепления гидроксила, т. е. по основному типу.
С ростом степени окисления увеличивается удельный заряд Э и преобладает диссоциация по кислотному типу с отщеплением иона водорода, так как связь Э—O упрочняется, а вследствие перераспределения электронной плотности у кислорода связь О—Н ослабевает. Таким образом, диссоциация по кислотному типу протекает, если ео–н << ЕЭ–О, а по основному типу – если ЕО–Н>> ЕЭ–О. При сравнимой прочности связей O—Н и Э—O диссоциация гидроокиси может одновременно протекать и по I и по II типам с отщеплением как гидроксила, так и иона водорода. Электролиты, которые в растворе ионизируются одновременно по кислотному и основному типам, называются амфотерными. Амфотерность в той или иной степени является общим свойством гидроокисей.
Количественно ионизация по тому или иному типу характеризуется константой ионизации:
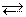 н+ + RO- roh R+ + он-
н+ + RO- roh R+ + он-
Kосн = [R+][OH-]/[ROH]
Kкисл = [RО-][Н+]/[ROH]
Из данных уравнений следует, что отношение константы ионизации по основному типу Kосн к константе ионизации по кислотному типу Ккисл равно
Kосн/Kкисл = [R+][OH-]/[RO-][H+]
Если отношение Kосн/Kкисл>>1, то ионизация в растворе идет преимущественно по основному типу, т.е. концентрация ионов ОН- во много раз превышает концентрацию гидратированных протонов Н+. Если же Kосн/Kкисл<<1, диссоциация протекает по кислотному типу. При Kосн/Kкисл» 1 диссоциация одновременно и в равной мере протекает по обоим типам. Например, для гидроокиси галлия Ga(OH)3 константы ионизации, соответствующие уравнениям
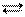 Ga(OH)3 Ga3+ + 3OH-
Ga(OH)3 Ga3+ + 3OH-
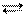 Ga(OH)3 3H+ + GaO33-
Ga(OH)3 3H+ + GaO33-
равны приблизительно 10-12 (т. е. Kосн = Kкисл). Следовательно, гидроокись галлия служит примером идеального амфолита (амфотерного электролита).
Применение принципа Ле-Шателье к кислотно-основному равновесию показывает, что с увеличением концентрации гидроксила возрастает вероятность ионизации по кислотному типу, а увеличение концентрации иона водорода приводит к преимущественной диссоциации по основному типу. Таким образом, в кислой среде амфолит проявляет основной, а в щелочной среде – кислотный характер. Например, гидроокись цинка при взаимодействии с кислотами ведет себя как основание:
Zn(ОН)2 + 2НС1 → ZnCl2 + 2Н2О
а при взаимодействии со щелочами – как кислота (H2ZnO2 или Zn(OH)2):
H2ZnO2 + 2NaOH → Na2ZnO2 + 2Н2О
Рассмотрим основные закономерности изменения характера ионизации гидроокиси в растворе в зависимости от положения элемента в Периодической системе. В ряду элементов III периода от натрия к хлору степень окисления растет, а эффективные ионные радиусы заметно уменьшаются. Ниже приведено изменение ионных радиусов элементов III периода в высшей степени окисления:
Элемент........ Na Mg Al Si P S Cl
Степень окисления... +1 +2 +3 +4 +5 +6 +7
Ионный радиус, Å... 0,98 0,78 0,57 0,39 0,34 0,29 0,26
В этом ряду резко возрастает объемная плотность заряда. Поэтому поляризующее действие ионов элементов, приводящее к перераспределению электронной плотности между связями Э—О и О—Н, возрастает в том же направлении. Гидроокиси крайних членов рассматриваемого ряда обладают чрезвычайно резко выраженными основными (NaOH) и кислотными (НС1O4) свойствами. Гидроокись натрия принадлежит к классу сильных растворимых оснований и является одной из самых сильных щелочей. Хлорная кислота по силе превышает такие кислоты, как НСl, H2SO4, HNO3 и др.
При переходе от натрия к магнию наблюдается некоторое ослабление основных свойств, однако Mg(OH)2 все же представляет собой довольно сильное основание. Гидроокись алюминия А1(ОН)3 уже является амфолитом с некоторым преобладанием основных свойств, а гидроокись кремния Si(OH)4 – амфолит с резко преобладающими кислотными свойствами и образует в растворе кислородсодержащие анионы. Последующие члены ряда обладают ярко выраженной склонностью к образованию в растворе сложных анионов, и соответствующие гидроокиси относятся к кислотам, причем их сила в ряду высших кислот Н2РO4 – H2SO4 – НС1O4 возрастает.
Для переходных металлов, образующих гидроокиси с переменной степенью окисления, характерны те же закономерности в изменении свойств. С возрастанием степени окисления и уменьшением при этом эффективного радиуса ионов ослабевают основные и нарастают кислотные свойства. В качестве примера рассмотрим ряд гидроокисей марганца Мn(ОН)2, Мn(ОН)3, Мn(ОН)4, Н2МnO4, НМnO4, в котором степень окисления марганца меняется в последовательности +2, +3, +4, +6, +7. Первые две гидроокиси – основания, последние две – кислоты, а гидроокись Mn(OH)4 – амфолит с некоторым преобладанием кислотных свойств.
Таким образом, если элемент образует гидроокиси в нескольких степенях окисления, то его гидроокиси в низших степенях окисления обладают более основным (или менее кислотным) характером, а гидроокиси в высших степенях окисления – более кислотным (или менее основным) характером.
|
|
|
|
|
Дата добавления: 2014-01-07; Просмотров: 1548; Нарушение авторских прав?; Мы поможем в написании вашей работы!