КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Величины квантового выхода некоторых фотохимических реакций
|
|
|
|
Квантовый выход фотохимической реакции
Под квантовым выходом - j фотохимических реакций понимается отношение числа прореагировавших молекул к числу поглощенных квантов:
j = число прореагировавших молекул/ число поглощенных квантов = m/n.
Опыт показывает, что далеко не во всех случаях j = 1. В большинстве случаев j < 1, но в ряде случаев j ³ 1. (Таблица 4).
Таблица 4.
| Реакция | j | l, нм | Светопоглощение вещества | Среда, в которой протекает реакция |
| 1. O2 + 2H2 ® 2H2O2 | 172-173 | O2 | Газовая среда | |
| 2. 2Fe2+ + I2 ® 2Fe3+ + 2I | I2 | Водный раствор | ||
| 3. 2HI ® H2 + I2 | 207-282 | HI | Газовая среда | |
| 4. 2HI ® H2 + I2 | 0,08-0,11 | 222-282 | HI | Водный раствор |
| 5. 3O2 ® 2O3 | 170-190 | O2 | Газовая среда | |
| 6. Co + Cl2 ® CoCl2 | 400-436 | Cl2 | Газовая среда | |
| 7. H2 + Cl2 ® 2HCl | 104-106 | 303-500 | Cl2 | Газовая среда |
| 8. CH3I ® CH4, C2H6, I2 | 0,01 | 202-210 | CH3I | Газовая среда |
Отклонение квантовых выходов от единицы можно объяснить следующим образом:
1. Возбужденные светом молекула или атом могут претерпевать не только фотохимические, но и фотофизические превращения – излучение кванта люминесценции или тепловую деградацию энергии. Сумма квантовых выходов всех этих первичных процессов åφi =1, следовательно, квантовый выход первичной фотохимической реакции может быть или равен, или меньше 1. Чем больше доля физических превращений, тем меньше квантовый выход j.
2. В большем случае продукты первичной реакции претерпевают дальнейшие изменения, т.е. за фотохимией, следует темновая стадия реакции. Например, образующиеся под действием света – радикалы А→R• + R•, могут соединиться обратно в молекулу А, а могут начать цепную реакцию.
Наличие темновых процессов приводит к двум важным следствиям:
1) квантовый выход реакции в целом может значительно отличаться от 1 как в большую, так и в меньшую сторону;
2) квантовые выходы одной и той же реакции могут существенно различаться в зависимости от того, при каких условиях и в какой сфере проводится реакция. Так, например, для реакции 2HI ® H2 + I2 в газовой сфере j=2, а в водном растворе j=0,8-0,11. Другой пример фотолиз кристаллов AgHal Þ при облучении осадка AgBr в водной среде актиничным светом (l = 400-420нм) j меньше 1 (j»0,2-0,3). Однако если к раствору над осадком добавить NaNO2 желатину или другой бромакцептор (т.е. вещество, поглощающее свободный бром), квантовый выход фотолиза AgBr ® Ag + Br поднимается почти до 1 (j»1). Бромакцептор связывает Br, выделяемый при фотолизе, и тем самым затрудняет (или делает невозможной) обратную реакцию образования AgBr: Ag + Br = AgBr. В отсутствие бромакцептора обратная реакция протекает в значительной степени, уничтожая результаты действия света, в силу чего j<1.
Квантовый выход меньше 1 может быть обусловлен не только обратной реакцией, но и другими причинами. Опыт показывает, что j<1 особенно часто наблюдается в случае реакций, протекающих в растворах, а также в газах при очень малых давлениях. В случае реакции в растворах j<1 обусловлен не только рекомбинацией, возникших при облучении продуктов реакции, но в значительной степени дезактивацией возбуждающих молекул молекулами-растворителями. В разреженных газах (а также в кристаллических телах при низких температурах), если реакция идет с участием возбуждающих молекул, М + hv = М*, активные молекулы, прежде чем они придут в контакт с другими молекулами, с которыми они должны прореагировать, дезактивируются путем самопроизвольного излучения поглощенной энергии, М* = М + hv.
В случае кристаллических тел при низких температурах потеря энергии возбуждения также идет путем люминесценции. В случае более концентрированных газов и при более высоких температурах кристаллических тел явление люминесценции играет меньшую роль, но возрастает вероятность рекомбинации продуктов реакций, а также потери энергии возбуждения активными молекулами в результате столкновения с аналогичными, но невозбужденными молекулами.
Фотохимические реакции протекают с квантовым выходом больше 1 тогда, когда темновые процессы, следующие за первичным, собственно фотохимическим актом, имеют то же, (но не обратное) направление, что и первичный акт. Например, реакция 2HI +hv ® H2 + I2 протекает в газовой среде с j=2. Это обусловлено следующей последовательностью реакций:
HI +hv ® H2 + I2 - первичный фотохимический акт, j=1;
Н• + HI ® H2 + I• - темновая реакция;
I• + I• ® I2 - темновая реакция;
_________________________________________________________
2HI +hv ® H2 + I2 – суммарная реакция, j=2.
В приведенных реакциях свободные атомы H и I обладают весьма высокой химической активностью, т.к. являются, по существу, свободными радикалами (отмечено точками). Особенно высокий квантовый выход наблюдается при протекании фотохимических цепных реакций. Типичный пример таких реакций:
H2 + Cl2 hν® 2HCl, j =104- 106.
Реакция протекает при облучении смеси Cl2 и H2 синим светом, что приводит к расщеплению молекул Cl2 на атомы: Cl2 + hv = 2Cl• (энергия квантов синей зоны недостаточна для расщепления молекулы Н2, но достаточна для диссоциации молекул Cl2). Образующиеся атомы Cl• химически весьма активны, поэтому при столкновении с молекулами H2 протекает реакция: H2 + Cl• ® HCl + H•. Образующиеся атомы H• реагируют с молекулами Н• + Cl2 ® HCl + Cl• далее цикл повторяется. Таким образом, поглощения первого кванта молекулой Cl2 ведет к расщеплению ее на атомы и развитию двух цепей темновых реакций с образованием HCl:
Cl2 + hv = Cl• + Cl• - первичный акт, j=1, зарождение цепей;
+Н2/ HCl+Н• +Н2/ HCl+Н• - темновые процессы, развитие цепей;
+Cl2/ HCl+ Cl• +Cl2/ HCl+ Cl•
+Н2/ HCl+Н• +Н2/ HCl+Н• - суммарное, j=104-106
Из приведенной схемы, казалось бы, следует, что достаточно поглощения одного кванта энергии для того, чтобы прореагировала вся смесь. В действительности, помимо развития цепей, имеет место их обрыв в результате реакции:
Н•+Н• ® Н2 + Q1 = 103,2ккал/моль; (1)
Cl•+ Cl• ® Cl2 + Q2 = 57,1ккал/моль;.(2)
где Q1, Q2 - теплоты соответствующих реакций Q1 = 103,2ккал/моль; Q2 = 57,1ккал/моль
Обрыв цепей особенно легко происходит на стенках сосуда, поэтому при прочих равных условиях, квантовый выход этой реакции тем больше, чем больше отношение объема сосуда к суммарной поверхности стенок. Обрыв цепей учащается также в случае, если к реагируемой смеси H2 + Cl2 добавлен какой-то газ, инертный по отношению к этой реакции (например, N2). Для сдвига реакций (1) и (2) в сторону молекул необходимо поглощение выделяемой энергии каким-то посторонним телом, что и выполняют стенки сосуда или молекула N2.
Знание квантового выхода очень важно для исследования механизма реакции и определения ее скорости.
Если заменить число поглощенных квантов и света на интенсивность поглощения света Ia, а число прореагировавших молекул на скорость изменения их концентрации – d[M]/dt, то получим выражение: - d[M]/dt = jl Iαl/Эl (Эl - величина энергии моля квантов). Из закона Бугера-Ламберта-Беера получим: d[M]/dt = jl I0l/Эl (I – 10-ε c l) выражение для скорости фотохимических реакций, объединяющее все законы фотохимии.
В заключении необходимо отметить следующее обстоятельство. Выше сформулирован закон квантовой эквивалентности Эйнштейна и указано, что число возбуждаемых молекул равно числу поглощенных квантов, на основании чего следует, что каждая молекула, поглощающая лучистую энергию, способна поглотить только один квант энергии. Это соотношение действительно соблюдается при обычных условиях проведения фотохимических реакций (квантовый выход первичного акта фотохимической реакции равен единице). Одноквантовость поглощения обусловлена тем, что продолжительность пребывания молекул в возбужденном состоянии очень мала (как правило, 10-8-10-9 с), а обычно используемые интенсивности облучения невысоки (1013 – 1015 квантов, поглощаемых 1 см3 за 1 с). поэтому концентрации возбужденных молекул малы и поглощение ими еще одного кванта маловероятно (аналогия с вероятностью тройного соударения молекул). Между тем, в настоящее время в связи с развитием техники получения потоков лучистой энергии весьма высокой интенсивности (излучение лазеров, техника импульсного фотолиза) стало возможным проведение двухквантовых процессов, т.е. таких фотохимических реакций, в которых молекула переходит в возбужденное состояние в результате поглощения не одного, а двух квантов энергии (так называемая двухквантовая фотохимия).
Основная литература (5 осн. [165-170])
Дополнительная литература (4 [18-32])
Контрольные вопросы:
1. Определение основного закона фотохимии
2. Понятие «квантовый выход фотохимической реакции»
3. Причины отклонения квантового выхода от единицы
4. Влияние условий и среды протекания фотохимической реакции на квантовый выход.
5. Причины понижения квантового выхода реакции (φ < 1).
6. Виды фотохимических цепных реакций.
7. Значение квантового выхода для определения скорости фотохимической реакции.
Лекция №4. Поглощение лучистой энергии атомами и молекулами
При поглощении атомами и молекулами ультрафиолетового (УФ) и видимого света происходят переходы электронов на более высокие энергетические уровни, в результате чего наблюдается типичный электронный спектр поглощения.
Поглощение света происходит лишь в случае, если разность между двумя энергетическими уровнями в точности соответствует энергии кванта:
Δ Е = h · ν = Е2 – Е1,
где Е2 и Е1 - энергия молекулы в конечном и начальном состоянии соответственно.
Если энергия начального состояния меньше энергии конечного Е1< Е2, то молекула или атом поглощают квант лучистой энергии. При обратном соотношении Е1 > Е2 квант излучается. Таким образом, возникают спектры поглощения или испускания. Явления, сюда относящиеся, известны также под названием поглощения, резонанса, флуоресценции, фосфоресценции одноатомных паров и газов и заключается в том, что данный газ, помещенный на пути пучка монохроматического света, частоту которого можно произвольно и непрерывно менять, при достижении ею некоторых определенных и характерных для элемента, составляющего газ, значений начинает испускать свет той же и других частот, а возбуждающий проходящий свет при этом ослабляется.
Молекулу вещества (например, красителя) в нормальном, или основном, состоянии с энергией Е0, обозначим через А, а молекулу, переведенную поглощением фотона hν на более высокий уровень энергии Е* назовем возбужденной и обозначим через А*.
В отличие от основного возбужденное состояние молекулы неустойчиво и имеет весьма краткую продолжительность существования. По истечении в среднем 10-9 с (и короче) большинство одновременно возбужденных молекул А* возвращается в исходное основное состояние и вновь может поглотить квант света. Многие красители, растворенные в жидких или твердых средах, поглощая свет, дают интенсивное свечение, не испытывая при этом никаких химических изменений. Наличие такого явления флуоресценции означает, что энергия, освобождаемая при возвращении возбужденной молекулы в основное состояние, излучается в виде фотона hνf. Рассматриваемые элементарные процессы поглощения и испускания световой энергии могут быть записаны в виде следующих физических реакций:
поглощение hνa + А → А*
испускание А* → А + hνf
Спектр испускания красителей представляет собой широкую полосу, занимающую в спектре, как и полоса поглощения, протяжение порядка 100 нм, но всегда сдвинутую по отношению к последней в сторону длинных волн. Такова закономерность явления флуоресценции, которая обнаружена и сформулирована под названием правила Стокса, согласно которому частота излучаемого света νf должна быть всегда меньше частоты νa света поглощенного:
hνf< hνa
Это означает, что в виде фотона всегда испускается только часть поглощенного кванта. Остаток hνa - hνf задерживается в молекуле в виде избыточной термической энергии, быстро отдаваемой окружению.
Функциональные группы, поглощающие электромагнитное излучение в УФ и видимой областях спектра называются хромофорами. Типичными хромофорами являются:
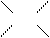 |
С=С (λ макс = 175;185 нм),  (λ макс = 160;185; 280 нм)
(λ макс = 160;185; 280 нм)
Функциональные группы (например, ОН, СООН, NH2, OR, NO2), которые вступают в сопряжение с хромоформами за счет своих неподеленных пар электронов с образованием новых хромофоров, называют ауксохромами.
Ультрафиолетовая и видимая спектроскопия дает информацию о наличии двойных или тройных углерод-углеродных связей, ароматических колец, азот-, кислород- и серосодержащих или других группировок, для которых характерны σ → σ*, π→π* и т.д. Обычно УФ и видимые спектры снимают в очень разбавленных растворах (10-4-10-6 моль/л), естественно в исследуемом диапазоне длин волн растворитель не должен иметь полосу поглощения.
При поглощении инфракрасного излучения (ИК-излучения) молекулы вещества переходят в колебательно-возбужденное состояние. Частоты колебаний атомов в молекуле, а, следовательно, и частоты поглощенного излучения, определяются природой атомов, их расположением в молекуле и характером связей. Так как молекулы разных веществ различаются по этим показателям, то и инфракрасные спектры сугубо характерны для каждого вещества.
Важной особенностью ИК-спектров является то, что группы атомов имеют характерное расположение полос поглощения в спектре, то есть определенные характеристические полосы. Поэтому по ИК-спектрам можно не только идентифицировать вещество, но и получить сведения о составе и строении неизвестного вещества.
В настоящее время определены характеристические частоты ИК-спектров многих веществ и групп атомов. Оказалось, что характеристические частоты групп атомов не имеют строго фиксированной величины, а лежат в довольно узких диапазонах, внутри которых обнаруживается поглощение этих групп в большом числе химических соединений.
Частоты излучений в ИК-спектроскопии принято выражать в волновых числах ω = 1/λ. Единицей измерения волнового числа является см-1. Волновое число пропорционально частоте
ω = ν/с, где ν – частота в с-1, с – скорость света в см/с. Полосы поглощения, наиболее характерные для молекулярных структур, лежат в интервале 5000-650 см-1, что соответствует длинам волн 2-15 мкм.
ИК-спектры можно использовать для качественного и количественного анализа веществ и их превращений, в том числе и для изучения фотохимических реакций. Строят спектральные кривые в координатах α % = f (ω) и τ % = f (ω). На кривых наблюдается система максимумов (пиков) поглощения, имеющих расположение, характерное для данного вещества. Пики поглощения соответствуют максимумам на кривых α % = f (ω) и минимумам на кривых τ % = f (ω). Волновые числа, соответствующие пикам поглощения называются характеристическими частотами. Характеристические частоты используются для качественного анализа вещества, а высота пиков – для количественного.
При качественном анализе, например, при идентификации неизвестного вещества, проводят сравнение его характеристических частот со справочными данными по спектрам поглощения различных функциональных групп. Если в спектре поглощения исследуемого неизвестного вещества есть максимумы в области характеристических частот данной функциональной группы, можно сделать заключение о наличии данной группы в молекуле. Необходимо учитывать, что наличие растворителя, образование водородных связей, кристаллизация могут привести к сдвигу максимума в спектре. Это затрудняет анализ неизвестного вещества и является достоинством при анализе вещества известного, так как позволяет получить о нем дополнительные сведения.
При фотохимических превращениях пики поглощения, характерные для реагента, начинают уменьшаться и при полном расходовании вещества исчезают. Пики, соответствующие продуктам реакции, наоборот, появляются и растут. Изменение спектров поглощения вещества позволяет судить об изменениях, происходящих в нем в процессе фотохимической реакции.
В таблице 5 приведены характеристические частоты некоторых функциональных групп фоторезистов, применяемых в полиграфической промышленности и электронной технике.
Таблица 5
Характеристические частоты химических групп
| Функциональная группа | Характеристическая частота, см-1 |
| Группа коричной кислоты в циннаматах -О-СО-СН=СНС6Н5 | |
| Группа >С=N2 в нафтохинондиазидах | 2100-2160 |
| Группа >С=О в нафтохинондиазидах | 1600-1620 |
| Группа -N≡N в бисазидах |
Высота пиков поглощения в спектре вещества (на спектральной кривой) определяется его количеством в системе. В случае соблюдения закона Бугера-Ламберта-Беера по высоте пика можно подсчитать концентрацию вещества, используя формулу: lg τλ = - ελ c l, где с – концентрация вещества в таблетке или пленке, ελ – молярный коэффициент поглощения, l – толщина образца.
Если исследуется фотохимическая реакция, происходящая в пленке, то концентрацию реагента или продукта в пленке в данный момент времени можно подсчитать по формуле:
Сх = С0 lg τх /τ0, где τ0 – коэффициент пропускания пленкой излучения с характеристической частотой для известной концентрации вещества, такой концентрацией может быть начальная С0;
τх - коэффициент пропускания для текущей концентрации (через время tх от начала реакции).
Так как по ИК-спектрам можно определить количество образовавшегося при реакции продукта, то их можно использовать для определения квантового выхода. По спектрам можно также рассчитать константу скорости фотолиза. Так как фотолиз о-нафтохинондиазида – реакция первого порядка, для него константу скорости фотолиза Кф можно определить по формуле:
Кф = d ln C/ dT. Перейдя по закону Бугера-Ламберта-Беера к оптической плотности, а затем к коэффициенту пропускания получаем Кф = Δln D/Δ t, где Δ t – интервал времени фотолиза, в течение которого высота пика изменилась с τ 0 τ х. Переход от τ к D осуществляется по формуле D = - lg τ
Основная литература (2 [21-33])
Дополнительная (5 [24-75])
Контрольные вопросы:
- Условия, необходимые для поглощения (испускания) света веществом
- Правило Стокса
- Понятие флуоресценции
- Определение понятий «хромофоры», «ауксохромы»
- Характеристические частоты в ИК-спектрах
- ИК-спектры для идентификации вещества
|
|
|
|
|
Дата добавления: 2014-11-16; Просмотров: 5042; Нарушение авторских прав?; Мы поможем в написании вашей работы!