КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Парасимпатическая нервная система 2 страница
|
|
|
|
Псевдогипопаратиреоз (см. также гл. 336). Генетический дефект, затрагивающий Гс-компонент аденилатциклазы, служит молекулярной основой для развития псевдогипопаратиреоза типа 1 (ПГП-1). Это редкое наследственное заболевание характеризуется гипокальциемией и гиперфосфатемией, повышенным уровнем содержания гормона паращитовидных желез в сыворотке крови (ГПЖ) и резистентностью к метаболическим эффектам вводимого экзогенного ГПЖ.
ГПЖ регулирует гомеостаз кальция, по меньшей мере частично, посредством стимуляции аденилатциклазы в почках и костях. Введение ГПЖ здоровым людям (и больным, страдающим идиопатическим или развившимся в результате хирургического вмешательства гипопаратиреозом) вызывает резкое увеличение экскреции циклического АМФ в мочу. В противоположность этому введение ГПЖ вызывает лишь небольшое увеличение (или вообще не изменяет) концентрации циклического АМФ в моче у больных, страдающих ПГП-1. Первоначально обнаружение этого факта породило предположение о том. что причиной ПГП-1 служит дефект в рецепторах ГПЖ, снижающий их способность стимулировать синтез циклического АМФ.
Однако влиянием дефекта, ограниченного только рецепторами ГПЖ, нельзя объяснить тот факт, что многие страдающие ПГП-1 больные также частично резистентны к действию других гормонов, включая тнреотропный (ТТГ), антидиуретический гормоны, глюкагон и гонадотропины. Действительно, некоторые страдающие ПГП-1 больные нуждаются в заместительной терапии по поводу симптоматического гипотиреоза вследствие резистентности щитовидной железы к действию ТТГ. Резистентность к действию других гормонов по большей части клинически не проявляется и ее можно выявить только с помощью специальных тестов.
То, что циклический АМФ служит для этих гормонов в качестве вторичного медиатора, позволяет предположить, что причиной ПГП-1 является дефект, действующий дистальнее гормональных рецепторов и изменяющий некий компонент, общий для всех опосредуемых циклическим АМФ реакций. Для многих людей, страдающих ПГП-1, такой компонент—Гс-белок, активность которого снижена в эритроцитах, тромбоцитах и фибробластах кожи у большинства больных приблизительно на 50%. У одного из больных было выявлено снижение активности Гс в почках, и, вероятно, аналогичные процессы наблюдались и в других клетках, являющихся эндокринными мишенями, включая кости, щитовидную железу, печень и т. д.
Возникает вопрос, если дефектность Гс — всеобщее явление при ПГП-1, то почему большинство из важных клинических последствий этого связано с резистентностью к единственному гормону — ГПЖ? Хотя на этот вопрос нельзя ответить определенно, вероятно, что большинство опосредуемых циклическим АМФ реакций на действие гормонов поддерживаются, несмотря на частичную неполноценность Гс, рядом компенсаторных механизмов, включая увеличение концентраций циркулирующих в крови стимулирующих гормонов. Однако наличие повышенной концентрации циркулирующего в крови ГПЖ оказывается недостаточным фактором для поддержания нормального гомеостаза кальция при ПГП-1, предположительно вследствие того, что нормальное функционирование ГПЖ решающим образом определяется условием нормальной активности Гс. К счастью, нарушения реактивности ГПЖ можно ликвидировать лечением витамином D, с помощью которого концентрации кальция и фосфора в сыворотке крови восстанавливаются до нормы.
Холера (см. также гл. 115). Повышение содержания циклического АМФ в клетках слизистой оболочки кишечника вызывает массивную секрецию воды и электролитов, что приводит к развитию холерной диареи. Патогенный Vibrio cholerae продуцирует белковый экзотоксин, способный стимулировать синтез циклического АМФ фактически во всех клетках организма. Клинически заболевание ограничивается слизистой оболочкой кишечника, поскольку токсин не абсорбируется из пищеварительного тракта, таким образом, другие ткани недоступны для токсина, вырабатываемого бактериями, находящимися в кишечнике.
В отличие от процесса стимуляции аденилатциклазы гормонами действие холерного токсина начинается постепенно и не прекращается немедленно после введения токсина. Причиной такого различия является то, что холерный токсин действует не посредством обратимого связывания со стимулирующим рецептором, а как фермент, образуя стабильную ковалентную модификацию компонента Гс-белка аденилатциклазы. После связывания токсина с клетками одна из его пептидных субъединиц проникает через клеточную мембрану, где катализирует АДФ-рибозилирование Гс, используя внутриклеточный никотинамидадениндинуклеотид (НАД) в качестве субстрата:
Гс + НАД+ ---токсин---> Гс-АДФ-рибоза + никотинамид + Н+.
АДФ-рибозилирование Гс увеличивает синтез циклического АМФ, по-видимому, путем снижения скорости гидролиза ГТФ в комплексе Гс—ГТФ-С, который синтезирует циклический АМФ (см. рис. 67-2).
Этот биохимический механизм помогает понять причину развития холерной диареи в результате жизнедеятельности относительно небольшого числа патогенных микроорганизмов, а также то, почему болезнь может продолжаться некоторое время и после ликвидации микроорганизмов из кишечника. Первый феномен объясняется тем, что данный токсин является ферментом и поэтому небольшого числа его молекул достаточно для АДФ-рибозилирования значительной части молекул Гс в клетке. Сохраняющийся повышенным уровень синтеза циклического АМФ после выведения токсина (по меньшей мере в экспериментальных исследованиях) коррелирует со стабильностью Гс-АДФ-рибозы. Большинство клеток, по-видимому, 'не содержат ферментов, способных отделить АДФ-рибозу от Гс, и поэтому действие токсина прекратится только тогда, когда молекулы Гс-АДФ-рибозы будут замещены заново синтезированными молекулами Гс. Для выздоровления может потребоваться даже замена самих клеток слизистой оболочки. Выяснение молекулярной основы действия холерного токсина послужило ценным инструментом обнаружения Гс и определения его характеристик, а также повысило уровень нашего знания биохимической основы действия гормонов.
Коклюш (см. также гл. 109). Молекулярный патогенез коклюша в. бронхах очень близок к патогенезу холеры в кишечнике. Ни один из этих двух микроорганизмов не внедряется в ткани, —оба они вызывают заболевание, вырабатывая экзотоксины, изменяющие синтез циклического АМФ в клетках хозяина. Bordetella pertussis секретирует два патогенных экзотоксина: молекулярной мишенью одного из них, называемого коклюшевым токсином, является связанный с гуаниннуклеотидом угнетающий белок аденилатциклазы. Другой токсин сам является молекулой аденилатциклазы.
Подобно холерному, коклюшевый токсин катализирует перенос АДФ-рибозы от НАД+ к белку мембраны; в данном случае это белок Ги. АДФ-рибозилированная форма Ги не способна взаимодействовать с Ри (см. рис. 67-2); в результате этого коклюшный токсин предотвращает угнетение аденилатциклазы ингибирующими лигандами, такими как мускариновые, a-адренергические и опиатные агонисты.
Ключевые бронхиальные клетки-мишени для коклюшного токсина и молекулярные события, связывающие АДФ-рибозилирование Г„ с симптомами коклюша, в значительной степени неизвестны. Однако реакция АДФ-рибозилирования помогает объяснить одно загадочное явление: лечение коклюшной инфекции антибиотиками в очень незначительной степени уменьшает тяжесть и длительность заболевания; и действительно, симптомы болезни могут сохраняться в течение трех и более недель после исчезновения микроорганизмов из трахеобронхиального дерева. АДФ-рибозилирование Ги (подобно АДФ-рибозилированию Гс, описанному выше) трудно обратимо с помощью клеточных ферментов. Вероятно, болезнь не отступит даже в отсутствие поступления дополнительных порций токсина, пока не будут замещены модифицированные белки Ги или клетки, содержащие такие белки.
Второй секретируемый В. pertussis патогенный продукт — аденилатциклазный токсин — остается ферментно инертным до тех пор, пока не внедрится в клетки хозяина. Там он активируется путем связывания с кальмодулином, в результате чего происходит существенное увеличение содержания циклического АМФ в клетке. В отличие от коклюшевого действие аденилатциклазного токсина быстро исчезает после удаления его источника; клеточные ферменты разрушают бактериальный фермент и концентрация циклического АМФ в клетке быстро возвращается к норме.
Хотя большинство связей между молекулярными действиями этих токсинов и развивающимся в результате этого заболеванием недостаточно ясны, эксперименты in vitro показывают, что оба токсина могут нарушать функцию нейтрофильных гранулоцитов человека—клеток, играющих основную роль в защите организма от инфекций. Накопление циклического АМФ, вызываемое аденилатциклазным токсином, нарушает способность нейтрофильных гранулоцитов убивать захваченные микроорганизмы. Коклюшный токсин путем АДФ-рибозилирования Ги или Ги-подобных молекул блокирует реакции нейтрофильных гранулоцитов (хемотаксис, высвобождение лизосомальных гидролаз и т. д.) на комплемент и другие факторы хемотоксина. Любой из этих двух эффектов или оба они могут способствовать увеличению чувствительности к легочным инфекциям, вызываемым другими микроорганизмами, что является частым осложнением коклюша.
Помимо их роли в качестве инструментов для исследования преобразования гормональных сигналов, знание этих токсинов может принести реальную пользу в практической медицине. Хотя современная практика иммунизации экстрактами целых коклюшевых бактерий эффективно предотвращает инфицирование, сама по себе иммунизация является причиной значительного числа случаев заболевания. Возбудители коклюша, специфически неспособные продуцировать любой из двух описанных выше токсинов, делаются безвредными для организма хозяина. В соответствии с этим иммунизация препаратами очищенного токсина должна предотвращать развитие болезни и снизить заболеваемость, вызываемую самой иммунизацией. Результаты предварительных клинических испытаний подкрепляют оба эти прогноза.
Сибирская язва (см. также гл. 98). Кожное инфицирование Bacillus antracis вызывает развитие характерного поражения с областью центрального некроза, окруженной выступающим подкожным отеком. В то время как некротические поражения, по-видимому, не связаны с участием системы циклического АМФ, «отечным фактором» в организме является аденилатциклаза. Подобно аденилатциклазному токсину коклюша отечный фактор получает доступ к клеткам хозяина, активируется клеточным кальмодулином и увеличивает внутриклеточную концентрацию циклического АМФ. Интересно отметить, что встречаются редкие случаи, когда у больных, в организм которых с пищей попали В. antracis, развилась водянистая диарея, неотличимая от такого же симптома холеры. Диарея в таких случаях, вероятно, является результатом проникновения отечного фактора внутрь клеток слизистой оболочки кишечника, приводящего к увеличению концентрации циклического АМФ и секреции соли и воды.
Циклический АМФ в клинической медицине. Большое число гормонов и медиаторов действуют путем стимуляции аденилатциклазы, а некоторые фармакологические антагонисты действуют путем блокирования их связывания со специфическими рецепторами — например, анаприлина с b-адренорецепторами и циметидина с На-гистаминовыми рецепторами. Терапевтическое действие этих веществ зависит от увеличения или уменьшения концентрации циклического АМФ в клетках-мишенях и тканях больных. Кроме этого, метилксантины (кофеин и теофиллин) блокируют фосфодиэстеразы циклических нуклеотидов и могут оказывать некоторые из своих лечебных воздействий (например, расширение бронхов) путем увеличения концентрации циклического АМФ в клетках.
В клинической практике определение содержания циклического АМФ в моче целесообразно при диагностике заболеваний, сопровождающихся нарушением гомеостаза кальция и концентрации ГПЖ. Значительная часть присутствующего в моче циклического АМФ образуется в клетках проксимальных канальцев, реагирующих на циркулирующий в крови ГПЖ. Таким образом, уровень содержания циклического АМФ в моче обеспечивает удобное «окно», заглянув в которое можно оценить действие ГПЖ на Почки и которое может отражать повышение концентрации ГПЖ (при гиперпаратиреозе), снижение ее (при гипопаратиреозе) или резистентность конечного органа к действию ГПЖ (при ПГП-1) (см. гл. 336).
Однако в настоящее время реальное значение циклического АМФ для медицины заключается в том, что он является инструментом для исследования и понимания нормальной и патологической регуляции, а также для разработки новых лекарственных средств. Образцы аденилатциклазы в настоящее время обычно используют для проверки новых веществ на их способность стимулировать или блокировать адренергические, гистаминергические и многие пептидные рецепторы. Гормональные рецепторы не являются единственными основными и специфическими точками контроля регуляции, опосредуемой через циклический АМФ; вероятно, что и другие белки (см. рис. 67-1) будут служить мишенями для новых лекарственных средств в будущем.
Прочие вторичные медиаторы. Хотя наиболее хорошо изученным гормональным вторичным медиатором является циклический АМФ, некоторые гормоны действуют посредством увеличения внутриклеточных концентраций других химических сигнальных веществ, включая ионы кальция и циклической гуанозин-3',5^монофосфат (циклический ГМФ). Например, определенные эффекты a-адренергических и холинергических (мускариновых) веществ, по-видимому, опосредуются повышенными концентрациями кальция в цитоплазме. Клетки многих типов содержат гуанилатциклазу, фосфодиэстеразы циклического ГМФ и белковые киназы, которые специфически стимулируются циклическим ГМФ. Тем не менее роль этого вторичного циклического нуклеотида в нормальной и патологической регуляции не совсем ясна.
ГЛАВА 68. МЕТАБОЛИТЫ АРАХИДОНОВОЙ КИСЛОТЫ И ИХ РОЛЬ В МЕДИЦИНЕ
Р. Поль Роберт сон (R. Paul Robertson)
Содержание этой главы сконцентрировано на образовании и механизме действия физиологически активных метаболитов арахидоновой кислоты и на биологическом феномене, в котором могут участвовать эти вещества.
Образование эйкосаноидов. Простагландины — первые из выделенных метаболитов арахидоновой кислоты — названы так потому, что впервые они были выявлены в сперме. Считалось, что они секретируются предстательной железой. По мере того как выявлялись другие активные метаболиты, становилось очевидным наличие двух основных путей их превращения — циклооксигеназного и липооксигеназного. Эти пути синтеза схематически представлены на рис. 68-1, а строение типичных метаболитов — на рис. 68-2. Все продукты как циклооксигеназного, так и липооксигеназного происхождения называют эйкосаноидами. Продукты циклооксигеназного пути — Простагландины и тромбоксан — простаноидами.
Начальный этап синтеза в обоих метаболических путях включает в себя отщепление арахндоновой кислоты от фосфолипида в плазматической мембране клеток. Затем свободная арахидоновая кислота может быть окислена циклооксигеназным или липооксигеназным путем. Первым продуктом циклооксигеназного пути является циклический эндопероксид простагландин G2 (ПГG2), который превращается в простагландин Н2 (ПГН2). ПГG2 и ПГН2 служат ключевыми посредниками в процессе образования физиологически активных простагландинов (ПГD2, ПГЕ2, ПГF2 и и ПГI2) и тромбоксана А2 (ТКА2). Первым продуктом 5-липооксигеназного пути является 5-гидропероксиэйкосатетраеноевая кислота (5-ГПЭТЕ), которая играет роль посредника при образовании 5-гидроксиэйкосатетраеноивой кислоты (5-ГЭТЕ) и лейкотриенов (ЛТА4, ЛТВ4, ЛТС4, ЛТD4 и ЛТE4). Две жирные кислоты, отличающиеся от арахидоновой кислоты, 3,11,14-эйкосатриеноивая кислота (дигомо-g-линоленовая кислота) и 5,8,11,14,17-эйкосапентаеновая кислота, могут превращаться в метаболиты. близкие по строению к этим эйкосаноидам. Простаноидные продукты первого субстрата обозначаются индексом 1; лейкотриеновые продукты этого субстрата—индексом 3. Простаноидные продукты второго субстрата имеют обозначение 3, в то время как лейкотриеновые продукты этого субстрата обозначаются индексом 5.
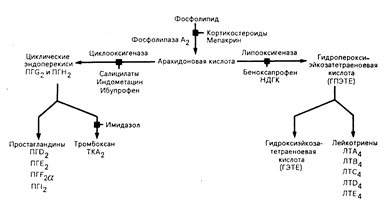
Рис. 68-1. Схема метаболизма арахидоновой кислоты. Различные лекарственные средства действуют на разные ферментные этапы, угнетая реакцию. Основными путями метаболизма являются циклооксигеназный и липооксигеназный. Фосфолипазу А2 угнетают кортикостероиды и мепакрин; циклооксигеназу — определенные салицилаты, индометацин и ибупрофен; липооксигеназу — беноксапрофен и нордигидрогуайаретиковая кислота (НДГК). Имидазол предотвращает синтез ТКА2.
Арахидоновая кислота образует простагландины, обозначаемые индексом 2, и лейкотриены, обозначаемые индексом 4. Индексы означают число двойных связей между атомами углерода в боковых цепях.
Фактически все клетки обладают необходимыми субстратами и ферментами для образования некоторых метаболитов арахидоновой кислоты, но различия ферментного состава тканей обусловливают различия в образуемых ими продуктах. Эйкосаноиды синтезируются по мере их непосредственной необходимости и не хранятся- в значительных количествах для последующего высвобождения.
Циклооксигеназные продукты. Простагландины D2, Е2, F2a и I2 образуются из циклических эндопероксидов ПГG2 и ПГH2. Из числа этих простагландинов ПГЕ2 и ПГI2 обладают наиболее широким спектром физиологического действия. ПГЕ2 оказывает заметное влияние внутри тканей и синтезируется многими из них. ПГI2 (называемый также простациклином) является основным продуктом арахидоновой кислоты в эндотелиальных и гладкомышечных клетках стенок сосудов и в некоторых несосудистых тканях. ПГI2 служит вазодилататором и угнетает агрегацию тромбоцитов. Считают, что ПГD2 также играет роль в агрегации тромбоцитов и функции головного мозга, а пгf2a — в функции матки и яичников.
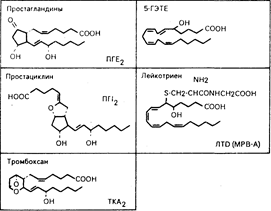
Рис. 68-2. Строение типичных биологически активных эйкосаноидов.
Тромбоксансинтетаза катализирует включение атома кислорода в кольцо эндоперекиси ПГН2 для образования тромбоксанов. TKA2 синтезируется тромбоцитами и усиливает агрегацию тромбоцитов.
Липооксигеназные продукты. Лейкотриены и ГЭТЕ являются конечными продуктами липооксигеназного пути. Лейкотриены обладают гистаминоподобным действием, включая индуцирование повышенной проницаемости сосудов и бронхоспазма, и, по-видимому, оказывают влияние на активность лейкоцитов. ЛТС4, ЛТD4 и ЛТE4 были идентифицированы как медленнореагирующие вещества анафилаксии (МРВ-А). (Патофизиология лейкотриенов детально обсуждается в гл. 202.)
Действие лекарственных средств на синтез эйкосаноидов. Многие лекарственные средства блокируют синтез эйкосаноидов путем угнетения одного или нескольких ферментов на путях их биосинтеза. Глюкокортикоиды и противомалярийные средства, такие как акрихин, препятствуют отщеплению арахидоновой кислоты от фосфолипидов (см. рис. 68-1). Циклооксигеназа непосредственно угнетается нестероидными противовоспалительными средствами, включая салицилаты, индометацин и ибупрофен. Беноксапрофен (Benoxaprofen)—еще одно нестероидное противовоспалительное средство — угнетает опосредуемое липооксигеназой превращение арахидоновой кислоты в ГПЭТЕ. Антидепрессант трансамин угнетает превращение циклических эндоперекисей в ПГI2, а имидазол — синтез тромбоксана. Тот факт, что какое-то лекарственное средство подавляет синтез определенного эйкосаноида, не означает, что действие данного лекарственного средства непосредственно приводит к дефициту этого продукта. Большинство этих лекарственных средств такого рода угнетают ранние этапы путей синтеза и поэтому блокируют образование не одного, а нескольких продуктов. Кроме того, некоторые из этих лекарственных средств оказывают и другие влияния. Например, индометацин не только угнетает образование циклических эндоперекисей, осуществляемое при помощи циклооксигеназы, но может также и нарушать транспорт кальция через мембраны, угнетать зависимые от циклического аденозинмонофосфата (циклического АМФ) протеинкиназу и фосфодиэстеразу, а также угнетать один из ферментов, ответственных за расщепление ПГЕ2. Не существует ни одного истинно специфичного ингибитора синтеза и ни одного специфичного антагониста рецепторов для отдельных метаболитов арахидоновой кислоты, которые можно было бы использовать в терапевтических целях. Отсутствие таких лекарственных средств является важным барьером, мешающим установить роль этих метаболитов в физиологических и патофизиологических процессах.
Метаболизм и количественный анализ эйкосаноидов. Метаболиты арахидоновой кислоты быстро диссеминируют in vivo. Простагландины серий Е и F, несмотря на то что они являются химически стабильными веществами, почти полностью расщепляются во время прохождения через печень или легкие. Таким образом, по существу все количество неметаболизированного ПГЕ2, определяемое в моче, образуется в результате секреции из почек и семенных пузырьков, в то время как содержащиеся в моче метаболиты ПГЕ2 характеризуют его синтез (ПГЕз) во всем организме. Как ПГI2, так и ТКА2 химически нестабильны и также подвергаются быстрой диссимиляции. Поскольку продолжительность жизни ПГЕ2, ПГI2 и ТКА2 in vivo невелика, измерение количества их неактивных метаболитов обычно используют в качестве показателя скорости их образования. ПГЕ2 превращается в 15-кето-13,14-дигидро-ПГЕ2; ПГI2 — в 6-кето-ПГF1a, а ТКА2 — в ТКВ2. Существует пять методов измерения содержания метаболитов арахидоновой кислоты в физиологических жидкостях: количественное определение биологической активности, радиоиммунный метод, хроматографический метод, определение числа рецепторов и масс-спектрометрия. При использовании любого из этих методов необходимо соблюдать определенные предосторожности при обращении с образцами биологических жидкостей, поскольку синтез простагландинов может быть повышенным во время сбора этих образцов. Например, если кровь свернулась или тромбоциты не были тщательно отделены от плазмы, то образование больших количеств ПГЕ2 и ТКА2 во время исследования может привести к получению ошибочных результатов. Добавление ингибитора синтеза простагландина в пробирку для сбора крови сведет эту проблему к минимуму.
Физиология. Простагландины и лейкотриены имеют специфические рецепторы на плазматических мембранах клеток печени, желтого тела, надпочечников, липоцитов, тимоцитов, матки, панкреатических островков, тромбоцитов и эритроцитов. Большинство этих рецепторов обладает специфичностью к эйкосаноидам определенного типа. Например, рецептор ПГЕ на плазматической мембране клеток печени связывает обладающие высоким сродством пге1 и ПГЕ2, но не связывает Простагландины классов A, F и I. Пострецепторные механизмы, с помощью которых связывание простагландинов изменяет функцию клетки, недостаточно ясны. Нормальное физиологическое функционирование эйкосаноидов не опосредуется через плазму крови. Вместо этого они действуют как местные, межклеточные и/или внутриклеточные модуляторы биохимической активности в тканях, в которых они образуются (например, пара.кринная функция). Эйкосаноиды являются аутокоидами, а не гормонами. Большинство из них обладает очень непродолжительным периодом жизни в циркулирующей крови вследствие их химической-нестабильности и/или быстрого расщепления.
Липолиз. ПГЕ2, синтезируемый липоцитами, имеет специфические рецепторы в липоцитах и является сильным эндогенным ингибитором липолиза. Поскольку для стимуляции липолиза гормонами необходимо образование циклического АМФ, было довольно подробно исследовано взаимодействие между ПГЕ и аденилатциклазой. ПГЕ угнетает липолиз путем снижения образования циклического АМФ в ответ на действие адреналина, адренокортикотропного гормона (АКТГ), глюкагона и тиреотропного гормона (ТТГ). Таким образом, ПГЕ может действовать как эндогенное антилиполитическое вещество, препятствуя стимуляции гормонами образования циклического АМФ.
Инсулин и ПГЕ могут действовать независимо друг от друга при их антилиполитическом воздействии на липоциты. Например, инсулин, но не ПГЕ, угнетает стимуляцию липолиза экзогенным циклическим АМФ в изолированных липоцитах, но оба эти вещества подавляют стимулированное гормоном образование циклического АМФ. Это позволяет предположить, что место действия инсулина находится дистальнее места стимуляции аденилатциклазы. В организме некоторых животных ПГЕ угнетает глюкагон-индуцированный липолиз, в то время как инсулин не оказывает влияния на этот процесс.
Баланс натрия и воды. Ренин-ангиотензин-альдостероновая система служит основным регулятором гомеостаза натрия, а контроль за водным балансом осуществляется главным образом вазопрессином. Метаболиты арахидоновой кислоты влияют на обе эти системы. ПГЕ2 и ПГI2 стимулируют секрецию ренина, а ингибиторы синтеза простагландинов оказывают противоположное действие. ПГЕ2 и ПГI2 уменьшают сопротивление почечных сосудов и увеличивают почечный кровоток; это приводит к перераспределению кровотока от наружного слоя коры почек к юкстамедуллярной области почек. Ингибиторы синтеза простагландина, такие как индометацин и меклофенамат (meclofenamate), напротив, уменьшают общий почечный кровоток и шунтируют оставшуюся его часть к наружному слою коры почек, что может привести к острому спазму сосудов почек и острой почечной недостаточности, особенно при уменьшении объема циркулирующей крови и отечных состояниях. ПГЕг является натрийуретиком, тогда как ингибиторы циклооксигеназы вызывают задержку натрия и воды в организме.
Индометацин также увеличивает чувствительность к экзогенному вазопрессину, например, у собак. И наоборот, ПГЕ2 уменьшает стимулированный вазопресеином транспорт воды. Поскольку такое действие ПГЕ2 нарушается введением дибутирилциклического АМФ, то наиболее вероятно, что ПГЕ2 будет препятствовать стимуляции аденилатциклазы вазопрессином.
Агрегация тромбоцитов. Тромбоциты обладают способностью синтезировать ПГЕ2, ПГD2 и ТКА2. Физиологическое значение ПГЕ2 и ПГD2 в функции тромбоцитов не установлено, ТКА2 является сильным стимулятором агрегации тромбоцитов; в противоположность этому ПГI2, образуемый в эндотелиоцитах стенок кровеносных сосудов, напротив, играет роль сильного антагониста агрегации тромбоцитов. ТКА2 и ПГI2 могут оказывать свои разнонаправленные воздействия, соответственно уменьшая и увеличивая образование циклического АМФ в тромбоцитах.
Противодействуют агрегации тромбоцитов ингибиторы синтеза эндогенных простагландинов. Например, единичная доза ацетилсалициловой кислоты может подавить нормальную агрегацию тромбоцитов на 48 ч и более, предположительно путем угнетения опосредуемого циклооксигеназой синтеза ТКА2. Длительность фазы угнетения циклооксигеназы единичной дозой этого препарата в тромбоцитах продолжительнее, чем в других тканях, поскольку тромбоцит в отличие от ядросодержащих клеток, способных синтезировать новые белки, не обладает соответствующими структурами для образования нового фермента. Следовательно, действие ацетилсалициловой кислоты продолжается до тех пор, пока не будут выделены в кровь вновь образованные тромбоциты. С другой стороны, эндотелиоциты быстро восстанавливают активность циклооксигеназы после прекращения лечения и, таким образом, восстанавливается продукция ПГI2. В этом заключается одна из причин того, что организм больных, принимающих ацетилсалициловую кислоту, не предрасположен к чрезмерному тромбообразованию. Кроме того, тромбоцит более чувствителен к препарату, чем эндотелиоцит.
Повреждение эндотелия может привести к агрегации тромбоцитов вдоль стенки кровеносного сосуда, вызывая местное уменьшение синтеза ПГI2 и тем самым открывая возможность избыточной агрегации тромбоцитов в месте повреждения сосудистой стенки.
Действие на сосуды. Вазоактивные свойства метаболитов арахидоновой кислоты относятся к числу самых замечательных эффектов этих веществ. ПГЕ2 и ПГI2 являются вазодилататорами, а ПГF2a, ТКА2 и ЛТС4, ЛТD4, ЛТE4 — вазоконстрикторами в большинстве участков сосудистого русла. Эти свойства, по-видимому, представляют собой результат их прямого действия на гладкую мускулатуру сосудистой стенки. Если системное артериальное давление поддерживается в пределах физиологической нормы, то действие расширяющих сосуды метаболитов арахидоновой кислоты приводит к увеличению кровотока. Однако в случае понижения артериального давления кровоток будет уменьшаться, поскольку при системной гипотензии индуцированное катехоламинами сужение сосудов скомпенсирует сосудорасширяющее действие простагландинов. Таким образом, при оценке влияния метаболитов арахидоновой кислоты на кровоток в сосудистом русле того или иного органа необходимо исключить существенные изменения системного артериального давления.
|
|
|
|
|
Дата добавления: 2014-11-20; Просмотров: 659; Нарушение авторских прав?; Мы поможем в написании вашей работы!