КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Принципы моделирования ионных кристаллов методом молекулярной динамики
|
|
|
|
Молекулярная динамика на графическом процессоре
Простейший алгоритм программы, реализующей моделирование ионных кристаллов (или других физических систем) методом молекулярной динамики, может быть представлен в форме, показанной на рис. 6.1.
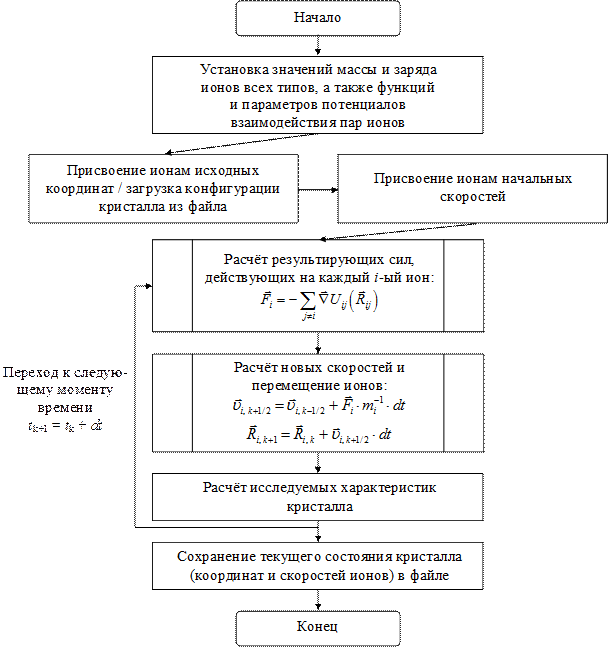
Рис. 6.1. Блок-схема алгоритма молекулярной динамики
Заключается этот алгоритм в том, кристалл представляется системой недеформируемых, положительных и отрицательных, ионов, эволюционирующей во времени. Ионы перемещаются по законам Ньютона, а силы взаимодействия определяются парными потенциалами Uij (Rij).
Потенциалы Uij (Rij) имеют общий вид
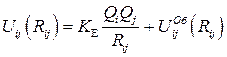 ,
,
где Qi, Qi, - заряды i -го и j -го ионов, R ij – расстояние между этими ионами, K E – константа закона Кулона, 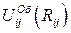 - некулоновский потенциал взаимодействия электроновских оболочек.
- некулоновский потенциал взаимодействия электроновских оболочек.
Потенциалы представляют в различных формах, обычно включающих в себя слагаемые, моделирующие отталкивание перекрывающихся электронных оболочек на малых расстояниях и дисперсионное притяжение на сравнительно больших. Наиболее известными из этих форм являются потенциал Букингема
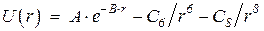
и потенциал Леннарда-Джонса
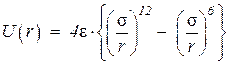 .
.
В формуле А и В – константы, характеризующие отталкивание оболочек, С 6, С 8 – константы, описывающие дипольную и квадрупольную составляющие дисперсионного притяжения. В формуле e - глубина минимума потенциала, а s - положение его нуля.
Перед началом расчёта ионам присваиваются некоторые начальные координаты и скорости (например - координаты, соответствующие узлам идеальной кристаллической решётки моделируемого соединения и скорости, соответствующие Максвелловскому распределению при заданной температуре), после чего начинается численное пошаговое интегрирование уравнений движения ионов во времени. На каждом k -ом шаге производятся следующие действия:
· Рассчитываются действующие на каждый ион силы
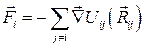 ;
;
· Вычисляются новые скорости и новые координаты ионов
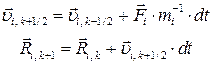 .
.
Формулы - справедливы при нулевых граничных условиях (конечный кристаллит из N частиц в вакууме) без компенсации перемещения, вращения и дрейфа температуры, возникающих из-за вычислительных погрешностей (алгоритмы компенсации приведены ниже). Однако этих формул достаточно, чтобы показать возможность эффективного распараллеливания по схеме SIMD: очевидно, что основные этапы алгоритма заключаются в проведении над каждым из ионов поочерёдно одних и тех же операций.
Наиболее критичным участком алгоритма является расчёт результирующих сил, действующих на каждый из ионов со стороны остальных. Этот расчёт необходим на каждом шаге молекулярной динамики, а его объём квадратичен по количеству частиц N, так как для каждой из N частиц необходимо выполнить суммирование сил, действующих со стороны N-1 остальных:
 .
.
Поскольку для реалистичного моделирования модельные кристаллиты должны содержать десятки и сотни тысяч ионов, объём расчёта очень велик по сравнению с расчётами на остальных этапах алгоритма. Именно этот расчёт имеет смысл в первую очередь реализовать на графических процессорах. Нам такая реализация позволила на порядки ускорить молекулярно-динамическое моделирование ионных кристаллов (диоксида урана).
6.2. Программирование графического процессора для расчёта действующих на ионы результирующих сил
Расчёт сил, действующих на ионы в молекулярной динамике, мы рассмотрим на конкретном примере нашего решения этой задачи. Структура исходных данных и алгоритм получения результата в данном случае несколько более сложны, чем в предыдущем примере сложения матриц. Прежде, чем переходить к анализу текстов программ, опишем подробнее исходные данные и ожидаемый результат.
|
|
|
|
|
Дата добавления: 2014-12-07; Просмотров: 550; Нарушение авторских прав?; Мы поможем в написании вашей работы!