КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
S- и р-элементы
|
|
|
|
Так, при переходе от s-элемента I группы к р -элементу VIII группы на кривой энергии ионизации атомов и кривой изменения их радиусов имеются внутренние максимумы и минимумы. Понятно, что экранирование ядра возрастает с увеличением числа внутренних электронных слоев. Поэтому в подгруппах s - и р -элементов наблюдается тенденция к уменьшению энергии ионизации атомов.
В характере изменения свойств s - и р -элементов в подгруппах отчетливо наблюдается вторичная периодичность (рис. 7). Для её объяснения привлекается представление о проникновении электронов к ядру. Как показано на рисунке 9, электрон любой орбитали определенное время находится в области, близкой к ядру. Иными словами, внешние электроны проникают к ядру через слои внутренних электронов. Как видно из рисунка 9, внешний 3 s -электрон атома натрия обладает весьма значительной вероятностью находиться вблизи ядра в области внутренних К - и L -электронных слоев.
Концентрация электронной плотности (степень проникновения электронов) при одном и том же главном квантовом числе наибольшая для s -электрона, меньше — для р -электрона, ещё меньше — для d -электрона и т. д. Например, при n = 3 степень проникновения убывает в последовательности 3 s >3 p >3 d, что эффект проникновения увеличивает прочность связи внешних электронов с ядром. Вследствие более глубокого проникновения s -электроны в большей степени экранируют ядро, чем р -электроны, а последние — сильнее, чем d -электроны.
Во внешнем слое у атомов d -элементов (за исключением Pd) находятся 1—2 электрона (ns -состояние). Остальные валентные электроны расположены в (n—1) d -состоянии, т. е. в предвнешнем слое.
Подобное строение электронных оболочек атомов определяет некоторые общие свойства d -элементов[18]. Так, их атомы характеризуются сравнительно невысокими значениями первой энергии ионизации. Как видно на рисунке 1, при этом характер изменения энергии ионизации атомов по периоду в ряду d -элементов более плавный, чем в ряду s - и p -элементов. При переходе от d -элемента III группы к d -элементу II группы значения энергии ионизации изменяются немонотонно. Так, на участке кривой (рис. 1) видны две площадки, соответствующие энергии ионизации атомов, в которых заполняются З d -орбитали по одному и по два электрона. Заполнение 3 d -орбиталей по одному электрону заканчивается у Mn (3d54s2), что отмечается некоторым повышением относительной устойчивости 4s2-конфигурации за счет проникновения 4s2-электронов под экран 3d5-конфигурации. Наибольшее значение энергии ионизации имеет Zn (3d104s2), что находится в соответствии с полным завершением З d -подслоя и стабилизацией электронной пары за счет проникновения под экран 3 d 10-конфигурации.
В подгруппах d -элементов значения энергии ионизации атомов в общем увеличиваются. Это можно объяснить эффектом проникновения электронов к ядру. Так, если у d -элементов 4-го периода внешние 4 s -электроны проникают под экран 3 d -электронов, то у элементов 6-го периода внешние 6 s -электроны проникают уже под двойной экран 5 d - и 4 f -электронов. Например:
| 22Ti …3d24s2 | I = 6,82 эВ |
| 40Zr …3d104s24p64d25s2 | I = 6,84 эВ |
| 72Hf… 4d104f145s25p65d26s2 | I = 7,5 эВ |
Поэтому у d -элементов 6-го периода внешние б s -электроны связаны с ядром более прочно и, следовательно, энергия ионизации атомов больше, чем у d -элементов 4-го периода.
Размеры атомов d -элементов являются промежуточными между размерами атомов s - и p -элементов данного периода. Изменение радиусов их атомов по периоду более плавное, чем для s - и p -элементов.
В подгруппах d -элементов радиусы атомов в общем увеличиваются. Важно отметить следующую особенность: увеличение атомных и ионных радиусов в подгруппах d -элементов в основном отвечает переходу от элемента 4-го к элементу 5-го периода. Соответствующие же радиусы атомов d -элементов 5-го и 6-го периодов данной подгруппы примерно одинаковы. Это объясняется тем, что увеличение радиусов за счет возрастания числа электронных слоев при переходе от 5-го к 6-му периоду компенсируется f -сжатием, вызванным заполнением электронами 4 f -подслоя у f -элементов 6-го периода. В этом случае f -сжатие называется лантаноидным. При аналогичных электронных конфигурациях внешних слоев и примерно одинаковых размерах атомов и ионов для d -элементов 5-го и 6-го периодов данной подгруппы характерна особая близость свойств.
Отмеченным закономерностям не подчиняются элементы подгруппы скандия. Для этой подгруппы типичны закономерности, характерные для соседних подгрупп s -элементов.
50. Получение высокомолекулярных соединений. Реакции полимеризации (ступенчатая и цепная полимеризация).
Природные ысокомолекулярные соединения, образующиеся в клетках живых организмов в результате биосинтеза, могут быть выделены из растительного и животного сырья с помощью экстрагирования, фракционного осаждения и других методов. Основные пути получения синтетических высокомолекулярных соединений - полимеризация иполиконденсация.
Карбоцепные высокомолекулярные соединения обычно синтезируют полимеризацией мономеров по кратным углерод-углеродным связям. Гетероцепные высокомолекулярные соединения получают поликонденсацией, а также полимеризацией мономеров по кратным гетероатомным связям типа С=О, N=C—О, С  N (например, альдегиды, изоцианаты, нитрилы) или с раскрытием гетероциклических группировок (например, окисей олефинов, лактамов).
N (например, альдегиды, изоцианаты, нитрилы) или с раскрытием гетероциклических группировок (например, окисей олефинов, лактамов).
Полимеризация – реакция образования полимера без образования низкомолекулярных продуктов. В качестве мономера используется молекула, содержащая кратную связь. При полимеризации этилена роль бифункциональной структурной единицы играет двойная связь, которая под влиянием инициатора (например, органического пероксида перикиси бензолоила (C6H5COO)2), легко переходит в радикальное состояние R∙; присоединение радикала создает условия для роста цепи:
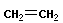 + + 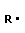 → → 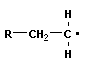
| инициирование | |
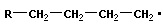
| рост цепи |
Для реакции полимеризации характерны три стадии: инициирование, рост цепи и обрыв цепи:
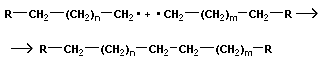
| обрыв цепи |
Этот тип полимеризации называется радикальным.
Полимеризация может инициироваться катионами или анионами (ионами). Ионная полимеризация включает те же стадии (инициирование, рост цепи, обрыв цепи). Инициаторами катионной полимеризации могут быть H+, неорганические апротонные кислоты SnCl4, AlCl3, металлоорганические соединения Al(C2H5)3. Инициаторами анионной полимеризации обычно служат электронодонорные соединения (щелочные металлы, их алкоголяты и т. д.).
Катионная полимеризация:
+ 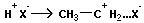
| ||
 + → + → 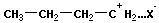
| и т. д. |
Анионная полимеризация:
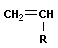 + +  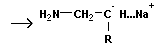
| ||
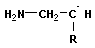 + + 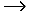
| ||
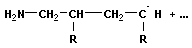
| рост цепи | |
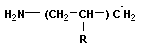 + + 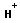 
| обрыв цепи |
Полимеризация может осуществляться между разными мономерами. Такие соединения называют сополимерами. В табл. 12.9 приведены примеры полимеров и сополимеров, получаемых реакцией полимеризации.
| ||||||||||||||||||
| Таблица 12.9 Важнейшие полимеры и сополимеры |
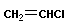
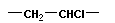
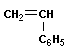

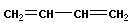
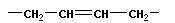
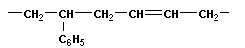
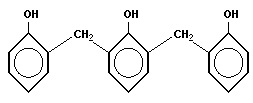
Поликонденсация сопровождается образованием полимера и низкомолекулярного соединения (H2O, HCl, NH3 и т. п.). Мономеры должны содержать минимум две функциональные группы.
Типичная реакция поликонденсации лежит в основе получения фенолформальдегидных смол
(n + 2) 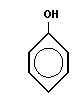
| + | n 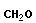
|
| 
| + | n 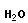
|
или полиэфирных соединений
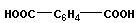 +
+ 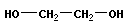
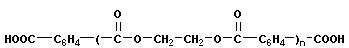 + n
+ n
В табл. 12.10 приведены наиболее типичные примеры, получаемые реакцией поликонденсации.
| ||||||||||||||||||
| Таблица 12.10 Важнейшие полимеры и сополимеры |
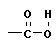
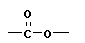
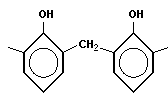
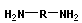
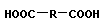 или
или 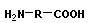
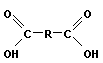
 +
+ 

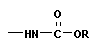 +
+ 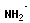
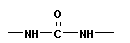
51. Полярные и неполярные молекулы. Дипольный момент связи и дипольный момент молекулы.
ПОЛЯРНЫЕМОЛЕКУЛЫ, молекулы, обладающие постоянным дипольным моментом в отсутствие внеш. электрич. поля. Дипольный момент присущ таким молекулам, у к-рых распределение электронного и ядерного зарядов не имеет центра симметрии. Обычно полярность отдельных фрагментов молекулы или хим. связей между двумя атомами (или большим числом атомов) определяется величиной соответствующего дипольного момента: чем он больше, тем сильнее полярность.
Под влиянием внеш. электрич. поля в-во поляризуется, т.е. в нем возникает дипольный момент единицы объема. У в-в, состоящих из полярных молекул, поляризация обусловлена смещением электронной плотности под влиянием поля и ориентацией молекул в поле. Ориентации молекул препятствует тепловое движение, поэтому изучение зависимости поляризации от т-ры позволяет определятьдипольный момент молекул (ур-ние Ланжевена-Дебая; см. Диэлектрики). Для двухатомных молекул полярность часто связывают с приближенным представлением электронной волновой ф-ции в рамках валентных связей метода как суммы двух слагаемых, одно из к-рых отвечает ковалентной схеме, другое -ионной валентной схеме. Такое соотнесение позволяет ввести понятие о степени ковалентности или степени ион-ности хим. связи, причем полярность связи определяется в осн. ионной составляющей. Для многоатомных молекул также возможно подобное приближенное выделение в электронной волновой ф-ции ковалентной и ионной составляющих.
Дипольный момент электрический, векторная величина, характеризующая асимметрию распределения положительных и отрицательных зарядов в электрически нейтральной системе. Два одинаковых по величине заряда +q и —q образуют электрический диполь с дипольный момент m = q l, где l - расстояние между зарядами. Для системы из n зарядов qi радиусы-векторы которых ri, 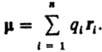 В молекулах и молекулярных системах центры положительных зарядов q А совпадают с положениями атомных ядер (радиусы-векторы r A), а электронное распределение описывается плотностью вероятности r(r).
В молекулах и молекулярных системах центры положительных зарядов q А совпадают с положениями атомных ядер (радиусы-векторы r A), а электронное распределение описывается плотностью вероятности r(r).
В этом случае дипольный момент 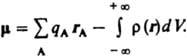 Вектор дипольный момент направлен от центра тяжести отрицательных зарядов к центру тяжести положительных. В хим. литературе дипольный момент молекулы иногда приписывают противоположное направление. Часто вводят представление о дипольный момент отдельных хим. связей, векторная сумма которых дает дипольный момент молекулы. При этом дипольный момент связи определяют двумя положительными зарядами ядер атомов, образующих связь, и распределением отрицательного (электронного) заряда.
Вектор дипольный момент направлен от центра тяжести отрицательных зарядов к центру тяжести положительных. В хим. литературе дипольный момент молекулы иногда приписывают противоположное направление. Часто вводят представление о дипольный момент отдельных хим. связей, векторная сумма которых дает дипольный момент молекулы. При этом дипольный момент связи определяют двумя положительными зарядами ядер атомов, образующих связь, и распределением отрицательного (электронного) заряда.
Дипольный момент химической связи обусловлен смещением электронного облака в сторону одного из атомов. Связь называют полярной, если соответствующий дипольный момент существенно отличается от нуля. Возможны случаи, когда отдельные связи в молекуле полярны. а суммарный дипольный момент молекулы равен нулю; такие молекулы наз. неполярными (напр., молекулы СО2 и CCl4). Если же дипольный момент молекулы отличен от нуля, молекула наз. полярной. Напр., молекула Н2О полярна; суммирование дипольных моментов двух полярных связей ОН также дает отличный от нуля дипольный момент, направленный по биссектрисе валентного угла НОН.
Порядок величины дипольный момент молекулы определяется произведением заряда электрона (1,6.10-19 Кл) на длину химической связи (порядка 10-10 м), т. е. составляет 10-29 Кл.м. В справочной литературе дипольный момент молекул приводят в дебаях (Д или D), по имени П. Дебая; 1 Д = 3,33564.10-30 Кл.м.
Спектроскопические методы определения дипольного момента молекул основаны на эффектах расщепления и сдвига спектральных линий в электрическом поле (эффект Штарка). Для линейных молекул и молекул типа симметричного волчка известны точные выражения, связывающие дипольный момент со штарковским расщеплением линийвращательных спектров. Этот метод дает наиб. точные значения величины дипольный момент (до 10-4 Д), причем экспериментально определяется не только величина, но и направление вектора дипольный момент Важно, что точность определения дипольный момент почти не зависит от его абсолютной величины. Это позволило получить весьма точные значения очень малых дипольный момент ряда молекул углеводородов. которые нельзя надежно определить другими методами. Так, дипольный момент пропана равен 0,085 b 0,001 Д, пропилена 0,364 b 0,002 Д, пропина 0,780 b 0,001 Д, толуола 0,375 b 0,01 Д, азулена 0,796 b 0,01 Д. Область применения метода микроволновой спектроскопии ограничена, однако, небольшими молекулами, не содержащими атомов тяжелых элементов. Направление вектора дипольный момент молекулы может быть определено экспериментально и по эффекту Зеемана второго порядка.
Другая группа методов определения дипольных моментов основана на измерениях диэлектрической проницаемости ε вещества. Этими методами измерены дипольные моменты молекул более 10 тыс. веществ. Переход от измеряемого значения ε газа, чистой жидкости или разбавленного раствора, то есть макроскопической характеристики диэлектрика, к величине дипольного момента основан на теории поляризации диэлектриков. Считается, что при наложении электрического поля на диэлектрик его полная поляризация Р (средний дипольный момент единицы объема) складывается из наведенной, или индуцированной, поляризации Р м и ориентационной поляризации Р ор и связана с m ур-нием Ланжевена - Дебая:
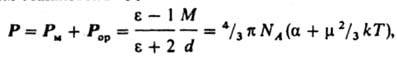
где М - мол. масса, d - плотность, a - поляризуемость молекулы, NA - число Авогадро, k - постоянная Больцмана, Т - абсолютная температура. Измерения диэлектрической проницаемости проводят в постоянном поле или при низких частотах, обеспечивающих полную ориентацию молекул по полю. При наиболее распространенном варианте метода - измерениях в разбавленных растворах неполярных растворителей - предполагается аддитивность поляризаций растворенного вещества и растворителя.
Сопоставление дипольных моментов полярных молекул некоторых органических соединений, полученных разными методами, показано в таблице.
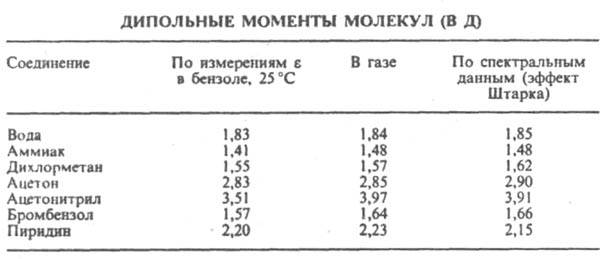
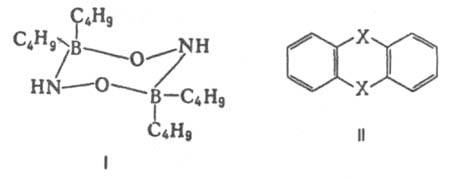
Важнейшая область применения данных о дипольных моментах молекул - структурные исследования, установление конформации молекул, конформационного и изомерного состава вещества, его зависимости от температуры. Величины дипольного момента молекул позволяют судить о распределении электронной плотности в молекулах и зависимости этого распределения от характера отдельных заместителей. В общем случае структурная интерпретация дипольный момент требует сравнения экспериментальных величин со значениями, полученными квантово-механическим расчетом либо при помощи аддитивной векторной схемы с использованием дипольных моментов отдельных связей и атомных групп. Последние находят либо по интенсивностям колебательных полос поглощения, либо путем векторного разложения дипольный момент некоторых симметричных молекул. Расчеты с использованием векторной аддитивной схемы могут учитывать различные проявления стереохимической нежесткости, например, затрудненное или свободной внутреннее вращение молекулы. Высокосимметричные молекулярные структуры, обладающие центром инверсии, двумя взаимно перпендикулярными осями вращения или осями, перпендикулярными плоскости симметрии, не должны иметь дипольный момент. По наличию или отсутствию дипольного момента молекулы можно в отдельных случаях выбрать для нее ту или иную структуру без каких-либо теоретических расчетов. Так, равенство нулю экспериментального дипольный момент димера аминооксидибутилборана (формула I) служит доказательством того, что он существует в виде устойчивой кресловидной конформации, обладающей центром инверсии. Наоборот, наличие дипольный момент у тиантрена (формула II, X = S) и селенантрена (II, X = Se), равных 1,57 Д и 1,41 Д соотв., исключает для них центросимметричную структуру, в частности плоскую.
52 – смотри 72
53. Понятие об электронных потенциалах. Уравнение Нернста.
Электродный потенциал, разность электрических потенциалов между электродом и находящимся с ним в контакте электролитом (чаще всего между металлом и раствором электролита). Возникновение Электродный потенциал обусловливается переносом заряженных частиц через границу раздела фаз, специфической адсорбцией ионов, а при наличии полярных молекул (в том числе молекул растворителя) — ориентационной адсорбцией их. Величина Электродный потенциал в неравновесном состоянии зависит как от природы и состава контактирующих фаз, так и от кинетических закономерностей электродных реакций на границе раздела фаз. Равновесное значение скачка потенциалов на границе раздела электрод/раствор определяется исключительно особенностями электродной реакции и не зависит от природы электрода и адсорбции на нём поверхностно-активных веществ. Эту абсолютную разность потенциалов между точками, находящимися в двух разных фазах, нельзя измерить экспериментально или рассчитать теоретически. Практическое значение имеют относительные Электродный потенциал, обычно называемые просто Электродный потенциал, представляющие собой разность Электродный потенциал рассматриваемого электрода и электрода сравнения — чаще всего нормального водородного электрода, Электродный потенциал которого условно принимается равным нулю.
При электрохимическом равновесии на электроде величина Электродный потенциал (E) может быть выражена через изменение гиббсовой энергии (D G) реакции: Е = — D G / zF, где z — число электронов, участвующих в электрохимическом процессе, F — Фарадея число. Электродный потенциал в этом случае зависит от активности (а) участвующих в реакции веществ (потенциалопределяющих веществ). Для электродов Me/Me n + Е = E 0 + (RT/zF) ln a Me n+, где R — газовая постоянная, Т — температура, E 0 — нормальный потенциал. Для окислительно-восстановительных систем с инертным электродом, у которых все компоненты электрохимической реакции находятся в растворе, Электродный потенциал (окислительно-восстановительный потенциал) определяется активностями как окисленной (a ok), так и восстановленной (а в) форм вещества.
Уравнение Нернста:
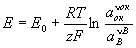 ,
,
где n — стехиометрический коэффициент.
В случае, когда на электроде возможно одновременное протекание более одной электродной реакции, используется понятие стационарного Электродный потенциал При пропускании электрического тока измеренный Электродный потенциал будет отличаться от равновесного на величину поляризации (см. Поляризация электрохимическая).
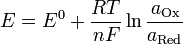 ,
,
где 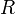 — универсальная газовая постоянная, равная 8.31 Дж/(моль·K);
— универсальная газовая постоянная, равная 8.31 Дж/(моль·K);
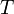 — абсолютная температура;
— абсолютная температура;
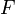 — число Фарадея, равное 96485,35 Кл/моль;
— число Фарадея, равное 96485,35 Кл/моль;
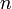 — число молей электронов, участвующих в процессе;
— число молей электронов, участвующих в процессе;
 и
и 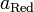 — активности соответственно окисленной и восстановленной форм вещества, участвующего в полуреакции.
— активности соответственно окисленной и восстановленной форм вещества, участвующего в полуреакции.
54. Последовательность заполнения электронных оболочек атомов. Принцип наименьших энергий. Принцип Паули. Правило Хунда.
Правило Хунда(Гунда) определяет порядок заполнения орбиталей определённого подслоя и формулируется следующим образом: суммарное значение спинового квантового числа электронов данного подслоя должно быть максимальным.
Это означает, что в каждой из орбиталей подслоя заполняется сначала один электрон, а только после исчерпания незаполненных орбиталей на эту орбиталь добавляется второй электрон. При этом на одной орбитали находятся два электрона с полуцелыми спинами противоположного знака, которые спариваются (образуют двухэлектронное облако) и, в результате, суммарный спин орбитали становится равным нулю.
При́нцип Па́ули (принцип запрета) — один из фундаментальных принципов квантовой механики, согласно которому два и более тождественных фермиона не могут одновременно находиться в одном квантовом состоянии.
Принцип был сформулирован для электронов Вольфгангом Паули в 1925 г. в процессе работы над квантомеханической интерпретациейаномального эффекта Зеемана и в дальнейшем распространён на все частицы с полуцелым спином. Полное обобщённое доказательство принципа было сделано им в 1940 г. в рамках релятивистской квантовой механики: волновая функция системы фермионов являетсяантисимметричной относительно их перестановок, поведение систем таких частиц описывается статистикой Ферми — Дирака.
Принцип Паули можно сформулировать следующим образом: в пределах одной квантовой системы в данном квантовом состоянии может находиться только одна частица, состояние другой должно отличаться хотя бы одним квантовым числом.
В статистической физике принцип Паули иногда формулируется в терминах чисел заполнения: в системе одинаковых частиц, описываемых антисимметричной волновой функцией, числа заполнения могут принимать лишь два значения Np = 0,1
55. Превращение химической энергии в электрическую.
Гальвани́ческий элеме́нт — химический источник электрического тока, названный в честь Луиджи Гальвани. Принцип действия гальванического элемента основан на взаимодействии двух металлов через электролит, приводящем к возникновению в замкнутой цепи электрического тока. ЭДС гальванического элемента зависит от материала электродов и состава электролита. Сейчас широко распространены следующие гальванические элементы:
| Тип | ЭДС (В) | Достоинства |
| угольно-цинковые (солевые) | 1,5 | дешёвые |
| щелочные (жаргонное название — алкалиновые) | 1,6 | высокий ток, ёмкие |
| никельоксигидроксидные (NiOOH) | 1,6 | высокий ток,очень ёмкие |
| литиевые | 3,0 | очень высокий ток, очень ёмкие |
Распространены солевые и щелочные элементы следующих типоразмеров:
| Американское название | Название МЭК | Название ГОСТ | Обиходное название |
| AAAA | R61 | ???? | ???? |
| AAA | R03 | мизинчик, тонкая | |
| AA | R6 | пальчик | |
| C | R14 | дюймовочка | |
| D | R20 | большая, бочка |
Распространены солевые и щелочные батареи элементов следующих типоразмеров:
| Название МЭК | Название ГОСТ | Обиходное название | Описание |
| 3R12 | квадратная, плоская | 3 элемента 12 (337) 4,5 В | |
| 6LR61 | _ | крона | 6 спец. галетных элементов 9 В |
В названии МЭК для щелочных элементов перед буквой R добавляется L, а для никельоксигидроксидных - буква X.
Также известны несколько десятков типоразмеров пуговичных (таблеточных) элементов разных электрохимических систем. Их обычно применяют в часах, микрокалькуляторах и других малогабаритных устройствах.
56. Предельные углеводороды. (см.46). Изомеры и гомологи.
Изомеры (от др.-греч. ἴσος — «равный», и μέρος — «доля, часть») — соединения (главным образом органические), одинаковые по элементному составу и молекулярной массе, но различные по физическим и химическим свойствам.
Гомологический ряд — ряд химических соединений одного структурного типа (например, алканы или алифатическиеспирты — спирты жирного ряда), отличающихся друг от друга по составу на определенное число повторяющихся структурных единиц — т. н. «гомологическую разность». Чаще всего это метиленовые звенья: …—СН2—… Простейший пример гомологического ряда — низшие гомологи алканов (общая формула СnH2n+2): метан CH4, этан C2H6, пропан С3H8и т. д.
57. Растворы полимеров. Растворимость и набухание полимеров.
|
|
|
|
|
Дата добавления: 2015-04-24; Просмотров: 1279; Нарушение авторских прав?; Мы поможем в написании вашей работы!