КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Вплив різноманітних факторів 2 страница
|
|
|
|
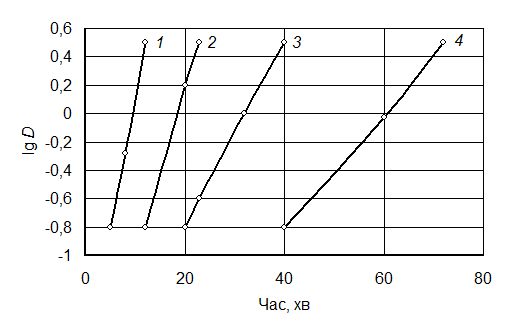
Рис. 4.7. Вплив розміру частинок вихідного оксиду
заліза (магнетиту) на тривалість відновлення:
1 – 25%; 2 – 50%; 3 – 75%; 4 – 100%
Збільшити газопроникність шихти в разі малого розміру частинок вихідних оксидів і тим самим збільшити швидкість відновлення можна застосуванням печей з обертовою трубою. Під час обертання труби (муфеля) відновлювальний матеріал пересипається в ній, тим самим у певні проміжки часу перебуває ніби в підвішеному стані, що забезпечує хороший контакт з газовим середовищем печі. У цьому разі максимальна температура відновлення обмежується стійкістю обладнання і економічними міркуваннями.
Вплив дифузії газів через прошарки шихти і продуктів відновлення як лімітуючої стадії відновлення можна виключити, створюючи для відновлення псевдозріджений стан порошкоподібних оксидів. Це можливо, відбувається, якщо відновлення в печах з киплячим шаром, у яких розміщена на перфорованих подах вертикального реактора шихта продувається знизу воднем під тиском 2,8…3,5 МПа. Швидкість подавання водню регулюється так, щоб частинки вихідних оксидів перебували в завислому стані. Так, можна отримувати порошок заліза, використовуючи як вихідну сировину порошкоподібний чи гранульований високозбагачений магнетит (99,7% Fe3O4 з 72% Fe) чи окалину. Відновлення при цьому проводиться за температури 540 °C.
Гранульовану шихту можна також застосовувати і за інших способів відновлення воднем. При цьому вигоряння у процесі відновлення зв’язків, використовуваних для грануляції, сприяє утворенню великих пор. Крім того, зберігається висока початкова пористість, зумовлена великим розміром гранул.
Вибір температури відновлення також може обумовлюватись вимогами до розміру частинок отримуваного порошку металу. Розмір частинок порошку металу найчастіше визначають розміром частинок вихідного чи нижчого оксиду, з якого безпосередньо утворюються частинки металу. Теоретично у процесі відновлення в оптимальних умовах з однієї частинки вихідного оксиду утворюється одна частинка порошку металу. При цьому розмір частинки металу завжди менший від розміру частинки оксиду. Однак на практиці не має однозначної залежності між розмірами. Особливо це характерно для відновлення металів, оксиди яких мають за високих температур підвищену пружність пари. У цих випадках застосування високих температур сприяє отриманню більших частинок металу, ніж це визначають вихідні розміри частинок оксидів. Для запобігання цьому явищу, незважаючи на прискорення процесу відновлення з підвищенням температури, її обмежують значеннями, що зменшують випаровування оксидів.
За певних умов можна отримувати дрібніші порошки металів, ніж це визначається вихідними оксидами.
Відзначені явища, характерні для відновлення оксидів металів воднем, зумовлені здебільшого перенесенням через газову фазу і топохімічними перетвореннями в твердій фазі. Класичний приклад щодо цього – процес отримання порошків вольфраму відновленням його оксиду WO3 воднем. Процес відновлення оксиду вольфраму відбувається через стадії утворення оксидів WO3–WO2,3–WO2,72 –WO2–W. Для отримання оптимальних розмірів частинок порошків вольфраму, що обумовлені розміром вихідних оксидів, процес проводять на стадії відновлення від WO3 до WO2 за температури 650...700 °С, а на стадії відновлення від WO2 до W – за температури 850…900 °С.
Під час проведення процесу на першій стадії за вищої температури, а також в умовах підвищеної вологості водню, створюються умови для отримання порошку вольфраму великого розміру. Це зумовлено тим, що вищі оксиди вольфраму WO3, WO2,3 і WO2,72 вже за температури 600…800 °С сублімують і утворюють з парою води сполуки WO3 · n H2О, WO2,9 · n H2О, які мають високу пружність пари. Випаровуючись, ці оксиди відновлюються на каталітично більш активних частинках знову утвореного оксиду WO2, збільшуючи їх, що зумовлює отримання більших частинок порошку вольфраму. За вищих температур можливе також випаровування оксиду WO2 у вигляді сполуки WO2 · n H2О з подальшим відновленням на частинках порошку вольфраму, що також збільшує їх розмір. Це явище використовують на практиці для отримання крупнозернистого порошку вольфраму. Для цього процес варто проводити за високої температури та швидкого її підвищення по довжині печі, підвищеної вологості водню і малої швидкості його подачі в робочий простір печі. Крім того, потрібно забезпечити високий прошарок WO3 у піддонах. Реально для отримання крупнозернистих порошків процес проводять на першій стадії за температури 800…900 °С і на другій – за 1200 °С чи на одній стадії за температури 1200 °С. Висока температура відновлення сприяє сублімації вищих оксидів WO3, WO2,9, а підвищена вологість газового середовища відновлення, що зумовлена малою подачею водню чи високим прошарком оксиду, – утворенню сполук WO3 · n H2О і WO2,9 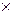 n H2О. Неодмінною умовою також є одночасне існування в робочому просторі вищих і нижчих оксидів WO3,WO2,9,WO2. Останнє досягається високим прошарком засипання вихідного оксиду і швидким підвищенням температури по довжині печі (швидким просуванням піддонів у високотемпературну зону печі) чи відновленням на одній стадії за температури 1200 °С. У цих умовах верхні прошарки засипки відновлюються до WO2 чи W раніше, ніж нижні, відновлення яких утруднюється через надходження водню до нижніх прошарків засипки і вилучення з них порів води дифузією по порових каналах.
n H2О. Неодмінною умовою також є одночасне існування в робочому просторі вищих і нижчих оксидів WO3,WO2,9,WO2. Останнє досягається високим прошарком засипання вихідного оксиду і швидким підвищенням температури по довжині печі (швидким просуванням піддонів у високотемпературну зону печі) чи відновленням на одній стадії за температури 1200 °С. У цих умовах верхні прошарки засипки відновлюються до WO2 чи W раніше, ніж нижні, відновлення яких утруднюється через надходження водню до нижніх прошарків засипки і вилучення з них порів води дифузією по порових каналах.
Велика товщина шару засипки може бути причиною збільшення частинок порошку вольфраму за рахунок проходження окиснювально-відновлювальних реакцій, що супроводжуються перенесенням через газову фазу. Це зумовлено тим, що коли шари засипки великої товщини чи підвищеної вологості водню, то в нижчих шарах шихти тиск пари води може наблизитись до рівноважного.
У цих умовах тиск пари поблизу частинок з малим радіусом кривизни, що визначається формулою Кельвіна – Томсона, може бути вищим, а поблизу великих – нижчим від рівноважного. Тоді у зв’язку з оборотністю реакції
WO2,9 + 0,9H2 = WO2 + 0,9H2О
дрібні частинки будуть окиснюватись до WO2,9, переходити у газову фазу у вигляді сполук WO2,9 · n H2О і відновлюватись на частинках WO2, тим самим збільшуючи їх.
Отримання крупних порошків перенесенням через газову фазу можливе тільки в тих випадках, коли можлива сумісність знаходження вищих і нижчих оксидів, тобто, якщо вони не утворюватимуть проміжних сплавів, розчинів і т. ін. Наприклад, отримання крупнозернистих порошків молібдену за дії механізму перенесення через газову фазу неможливе, не зважаючи на високу пружність парів оксиду молібдену МоО3. Таку пружність зумовлено тим, що оксид молібдену МоО3 утворює з проміжним оксидом складу Мо4О11 евтектику з температурою плавлення близько 550 °С. Таким чином, не виконується одна з умов отримання великих частинок – сумісне знаходження нижчого і вищого оксидів за високої температури (температура отримання порошку молібдену 900…950 °С). Тому на першій стадії нагрівання відновлюють повільно до температури 500 °С, забезпечуючи тим самим повне відновлення МоО3 до МоО2. Потім температуру підвищують до 900…950 °С. Інакше, коли оксид попадає у високотемпературну зону, шихта плавиться.
Розглянуті особливості відновлення оксидів молібдену зумовлюють малий розмір частинок порошку молібдену.
Отримання порошків металів і сплавів з малим розміром частинок іноді є метою технологічного процесу, що розроблюється. У цьому випадку відповідальними за формування таких частинок є кристалохімічні і топохімічні перетворення. Сутність їх полягає в тому, що за відновлення в оптимальних умовах з вихідної частинки вищого оксиду утворюється одна чи декілька частинок нижчого оксиду (рис. 4.8, а). Якщо на стадії утворення нижчого оксиду створити умови, за яких виникає декілька центрів зародження нової фази, то з однієї частинки вихідної фази утворюється декілька частинок нової, що й приводить до отримання дрібнішого продукту (рис. 4.8, б).
До переліку чинників, які сприяють отриманню дисперсних порошків, варто віднести такі, які забезпечували б вищу швидкість зародження нової фази порівняно з її ростом: використання вихідних матеріалів з підвищеною питомою поверхнею; швидке нагрівання на стадії зародження нової фази; проведення реакцій в умовах, далеких від рівноваги; швидке вилучення продуктів реакції, відсутність їх у відновлювальному середовищі (водні). Наявність продуктів реакції у відновлювальному середовищі сприяє проходженню процесів поперемінного окиснення – відновлення і утворенню летких сполук, що приводять до збільшення кінцевого металевого продукту. Крім того, підвищення концентрації пари води з високою адсорбційною здатністю посилює їх адсорбцію на найбільш активних місцях поверхні, у зв’язку з чим кількість зародків нової фази зменшується.
Знаючи вплив наведених чинників, а також фазовий склад у системах Ме-О і їх властивості, можна регулювати розмір частинок отримуваних порошків.
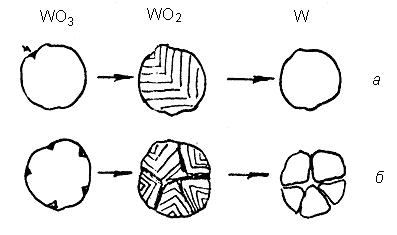
Рис. 4.8. Схема перетворень у процесі відновлення оксидів із застосуванням стандартних режимів (а) і таких, що забезпечують отримання
дисперсних порошків (б)
Вуглець застосовують для відновлення важко- і легковідновлювальних оксидів, якщо в кінцевому продукті допустимий деякий вміст вуглецю у вільному чи зв’язаному вигляді.
Раціональні умови проведення процесів відновлення металів з їх оксидів вуглецем значною мірою визначаються механізмом цих процесів. Варто відзначити, що взаємодія оксидів металів з вуглецем – найскладніший варіант відновлювального процесу, оскільки в його основі можуть існувати різні перетворення: контактна взаємодія оксиду і вуглецю з дифузією вуглецю до оксиду через прошарок металу, що утворюється, і газоподібних продуктів відновлення у зворотному напрямі, випаровування оксидів з їх відновленням на поверхні каталітично активніших частинок вуглецю (як правило, сажі); відновлення оксидів вуглецем, дисоціація оксиду на метал чи нижчий оксид і кисень, який потім окиснює вуглець з утворенням оксиду вуглецю. Останній надалі бере участь у процесі відновлення:
2МеО = Ме + О2;3С + 2О2 = СО2 + 2СО.
Однак найбільш імовірний механізм відновлення оксидів, особливо за високих температур, за яким спочатку відбувається газифікація вуглецю за реакціями
С + О2 = СО2 і СО2 + С = 2СО,
а потім відновлення оксиду металу оксидом вуглецю:
МеО + СО = Ме + СО2 .
У цьому випадку константу рівноваги реакції відновлення можна визначати так:
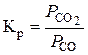 .
.
Рівновагу реакції відновлення варто розглядати у зв’язку з рівновагою реакції газифікації вуглецю. Наприклад, для випадку відновлення заліза і вольфраму рівновага реакцій відновлення нижчих оксидів FeO і WО2 має вигляд, як на рис. 4.9. Як видно з рисунка, відновлення оксиду заліза FeO нижче за температуру 650 °С й оксиду вольфраму нижче від 730 °С оксидом вуглецю неможливе, оскільки нижче від цих температур відбувається зворотна реакція:
2СО ® СО2 + С,
і рівноважна концентрація СО не досягається.
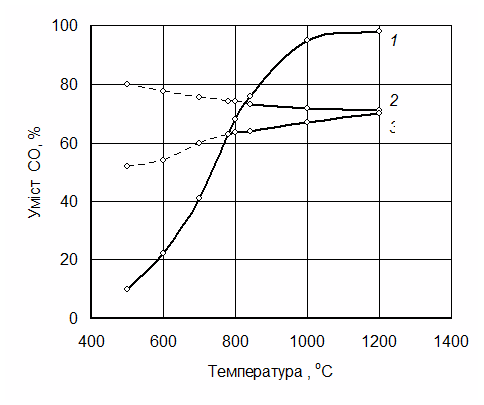
Рис. 4.9. Криві рівноваги реакцій:
1 – С + СО2 = 2СО; 2 – WO2 + CO = W + CO2;
3 – FeО + CO = Fe + CO2
Точки перетину кривих 3 і 2 з кривою 1 на рис. 4.9 рівноважні для реакцій відповідно:
FeO + CO = Fe + СО2і WО2 + 2СО = W + 2СО2 .
У разі проведення процесу за температури, нижчої від температури рівноваги відновлення, може відбуватись тільки за безпосередньої взаємодії оксиду з вуглецем, а за високої пружності пари оксиду, що відновлюється, – у разі відновлення його на частинках вуглецю. Однак відновлення за низьких температур відбувається повільно. Тому процес віднов-лення вуглецем зазвичай проводять за температур, вищих за температури рівноваги, за яких забезпечується необхідна концентрація СО, що визначається константою рівноваги реакції. Звичайно, ці температури вищі за 1000 °С, за яких газова фаза майже повністю складається з оксиду вуглецю. Процес можна прискорювати і за вищих температур, якщо це не стає причиною погіршення газопроникності вихідної шихти через спікання відновлених частинок металу.
Застосування вищих температур для відновлення вуглецем не сприяє збільшенню частинок порошку металу навіть, коли вихідні оксиди мають високу пружність парів. У цьому випадку за наявності дисперсних частинок сажі пари відновлюються не на частинках нижчих оксидів, а на каталітично активніших частинках сажі. Крім того, відсутність в газовому середовищі пара води приводить до менш інтенсивного випаровування вищих оксидів у вигляді сполук МеО · n H2О, як у розглянутому випадку відновлення оксиду вольфраму WО3 воднем.
Відновлення вуглецем передбачає отримання крупних порошків металів за температур, за яких отриманий порошок металу спікається в конгломерати. Надалі конгломерати подрібнюють механічними методами з потрібним ступенем подрібнення.
Оскільки в процесі відновлення вуглецем у реакції бере участь газоподібний компонент, то цей процес розвивається відповідно до закономірностей адсорбційно-автокаталітичної теорії, тобто лімітуючими стадіями процесу відновлення можуть бути фізична адсорбція і хемосорбція, хімічна реакція, дифузія газу-відновника і газоподібних продуктів відновлення по порових каналах, дифузія в конденсованій фазі. У цьому разі кінетика відновлення подібна до кінетики відновлення газоподібним відновником (Н2, СО).
У разі відновлення вуглецем лімітуючу стадію процесу можна визначити механізмом відновлення, який залежить від властивостей оксиду і металу, а також умов його отримання. Зазвичай можуть діяти одночасно декілька механізмів, причому частка кожного в загальному процесі відновлення металу змінюється зі зміною умов і ступеня відновлення.
Якщо швидкість процесу лімітується реакцією відновлення оксидів металів оксидом вуглецю, важливим є прискорення процесу газифікації вуглецем. Це можна забезпечити застосуванням для відновлення сажистого заліза чи введенням у вихідну шихту домішків солей деяких лужних металів.
Сажисте залізо являє собою синтетичний залізовуглецевий матеріал, який отримують термокаталітичним розкладанням вуглеводів (природного газу) на залізній губці чи в процесі їх взаємодії із залізнорудним матеріалом за температури 700…900 °С. Отриманий таким чином продукт – конгломерат частинок заліза і вуглецю з міцним зв’язком між ними і вмістом вуглецю 25…50%.
Застосування сажистого заліза у складі твердого чи комбінованого відновника для отримання порошків заліза збільшує швидкість відновлення і дозволяє знизити температуру процесу на 100…200 °С, оскільки вуглець у сажистому залізі знаходиться в активному стані внаслідок високого ступеня неупорядкованості кристалічної структури. У зв’язку з цим енергія активації процесів окиснення вуглецю сажистого заліза –
29 кДж/моль, а взаємодії його з вуглекислим газом (СО2) – 13 кДж/моль. При цьому швидкість процесу в інтервалі температур 680…800 °С можна визначити за рівнянням, мг/хв:
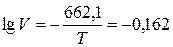 ,
,
де Т – температура, К.
У той же час збільшення ступеня упорядкованості кристалічної будови для пічної і лампової сажі, графіту приводить до збільшення енергії активації процесу їх газифікації відповідно до 280,2, 287,0 і
378,0 кДж/моль. Зменшення енергії активації процесів взаємодії вуглецевих матеріалів у міру збільшення розупорядкованості їх кристалічної структури з газоподібними окиснювальними компонентами зумовлено тим, що ці матеріали мають слабкий зв’язок поверхневих атомів і великі швидкості міграції атомів вуглецю. Реакція взаємодії вуглецю у сажистому залізі з О2, СО2 і Н2О лімітується надходженням газоподібних компонентів з пічного простору до реакційної поверхні.
Крім зазначених чинників, на швидкість газифікації сажистого заліза, як і на інші види вуглецевої сировини, впливають його дисперсність, пористість, питома поверхня, газопроникність вихідної шихти в цілому.
Активуючу роль домішок солей деяких лужних металів (наприклад, карбонатів і хлоридів) у процесах відновлення таких металів, як свинець, цинк, нікель, залізо, кобальт, мідь, марганець, ніобій та інші, зумовлено здебільшого двома причинами: 1) іони лужного металу, упроваджуючись у міжбазисні простори кристалічної гратки вуглецю, сприяють прискоренню його газифікації; 2) ті ж іони лужних металів взаємодіють з поверхнею оксидів металів і прискорюють структурні зміни в процесі їх кристалохімічних перетворень. Так, у разі хемосорбції на поверхні відновлювальної сполуки атомів більш важких лужних металів з меншим іонізаційним потенціалом активно порушується стан рівноваги системи в результаті локального порушення електронейтральності, що виводить її зі стану електронної рівноваги. До встановлення цієї рівноваги система прагне по-різному, зокрема, змінюючи валентність основних іонів, що утворюють гратку, тобто створюючи електронні вакансії.
Електронний обмін відбувається між відновником й оксидом, і процес в цілому інтенсифікується. Каталітична дія лужних металів посилюється з переходом від легших до більш важких металів.
Уведення солей лужних металів до складу шихти для відновлення покладено в основу ‹‹содового›› способу отримання залізного порошку. Додавання в цьому та інших випадках 10…20% соди (Na2CO3) поряд з указаним позитивним впливом на процес відновлення чинить також рафінуючу дію, оскільки зв’язує домішки, що містяться у вихідній сировині (силіцій, алюміній, оксиди силіцію, алюмінію, магнію, кальцію, титану, ванадію, хрому, молібдену, вольфраму та інших металів), у розчинні силікати типу Na2SiO3, NaCr2O3, які потім легко вилучаються гідрометалургійною обробкою водою чи розведеними кислотами подрібненого відновленого продукту.
У разі відновлення вуглецем і вуглецевмісними газами відбувається насичення вуглецем відновленого металу з утворенням твердих розчинів вуглецю в металі чи карбідів. Цей процес небажаний, оскільки підвищена твердість отримуваних порошків у цьому випадку знижує їх пресованість, сприяє підвищеному зношенню прес-форм. Для запобігання цим явищам відновлення проводять за можливо низьких температур, скорочують час перебування шихти в реакційній зоні. Але найдієвішим чинником щодо цього, що не знижує продуктивність пічного обладнання, є відновлення у суміші газів Н2 і СО. При цьому намагаються забезпечити в реакційному просторі мінімальну кількість газів CO2 і Н2О. Робоче газове середовище із суміші Н2 і СО створюється штучно або введеням водню в муфель печі для відновлення оксидів вуглецем. У результаті в процесі відновлення беруть участь гази Н2, СО, Н2О і СО2. У цьому випадку поряд з реакцією регенерації оксиду вуглецю
СО2+ С – 2СО
можуть відбуватись реакції:
С + Н2О = Н2 + СО;
СО2 + Н2 = Н2О + СО. (4.6)
Константа рівноваги реакції (4.6) визначається із співвідношення:
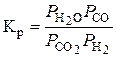 .
.
Оскільки вміст СО і СО2 визначається константою рівноваги для реакції регенерації вуглецю
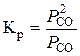 ,
,
вміст газів Н2, СО, СО2, Н2О у робочому просторі за певної температури і тиску чітко визначено. Наприклад, у разі відновлення оксидів заліза за температури нижчої за 818 °С, рівноважний вміст водню у газовій фазі (64%) більший від рівноважного вмісту СО (62,5%), а за температури вищої за 818 °С навпаки (60,8% Н2 і 67,1% СО) відновлювальна здатність водню вища, ніж оксиду вуглецю. У зв’язку з цим, а також через те, що коефіцієнт дифузії водню в газовому середовищі значно вищий, ніж у СО, для відновлення за температури понад 900 °С потрібне переважання в газовій суміші водню і оксиду вуглецю водню. Для цього найбільш прийнятний конвертований газ (75% Н2+25% СО), отриманий парогазовою конверсією природного газу за наявності нікелевого каталізатора за температури 1000…1100 °С. Утворення Н2 і СО відбувається за реакцією
СН4+ Н2О - 3Н2+СО.
Кінетика процесів відновлення оксидів металів вуглецем багато в чому визначається лімітуючими механізмами реакцій відновлення, основним з яких є дифузія газоподібних складових реакції – оксиду вуглецю, парів оксидів металів чи продуктів їх дисоціації, а також газоподібних продуктів реакції в газовій фазі в порових каналах та конденсованій фазі. Наприклад, у разі відновлення оксидів заліза за методом ЦНІІ Чермет і Сулінського заводу основну шихту, що складається з оксидної сировини і вуглецю, завантажують у робочий простір прошарками, що не перемішуються. Кінетика переміщення меж між прошарками описується параболічною залежністю
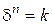
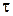 ,
,
де 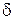 – товщина відновленого прошарка шихти; – час відновлення; n – показник, що залежить від характеристики шихти й умов процесу.
– товщина відновленого прошарка шихти; – час відновлення; n – показник, що залежить від характеристики шихти й умов процесу.
Такий закон росту відновленого шару свідчить про те, що лімітуючою стадією, що визначає в цьому випадку швидкість відновлення, є дифузія оксиду вуглецю через прошарок відновленого продукту:
СО2 + С=2СО.
Сумарна швидкість процесу відновлення залежить від швидкості дифузії СО і у зворотному напрямі СО2 через порові канали відновленого шару. У міру наростання цього шару і спікання відновлених частинок заліза опір дифузії зростає. У загальному випадку кінетика відновлення оксидів металів вуглецем, коли лімітуючою стадією є реакція генерації СО, участь його в процесі визначається здебільшого тими ж закономірностями, що й процеси відновлення оксидів металів воднем.
Коли в процесі відновлення істотну роль відіграє випаровування оксидів з подальшим їх перенесенням через газову фазу і відновленням на частинках вуглецю (сажі) важливого значення набувають швидкості випаровування і масоперенесення оксиду металу. Як зазначає О. П. Колцін, дійсна швидкість випаровування оксиду в реальних умовах може бути визначена з виразу
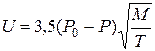 , (4.7)
, (4.7)
де Р 0 – рівноважний тиск пари; Р – парціальний тиск пари над поверхнею оксиду; М – молекулярна маса; Т – температура, оС.
Таким чином, дійсна швидкість випаровування тим менша, чим більше парціальний тиск пари оксиду наближений до рівноважного. При цьому істотне значення мають градієнт тиску пару оксиду, тиск оксиду вуглецю і відстань між поверхнями частинок оксиду і вуглецю.
Якщо не враховувати тиск оксиду вуглецю, що може бути в процесі низькотемпературного відновлення чи слабкого перебігу реакції газифікації вуглецю, швидкість випаровування оксиду можна визначити так:
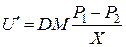 , (4.8)
, (4.8)
де D – коефіцієнт дифузії парів оксиду; Р 1, Р 2 – парціальний тиск парів оксиду над поверхнею оксиду чи відновника; Х – ефективна відстань між поверхнями оксиду і вуглецю.
Під час відновлення оксидів металів вуглецем тиск оксиду вуглецю часто дуже високий і нехтувати ним не можна. У цьому разі швидкість випаровування
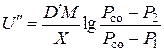 , (4.9)
, (4.9)
де  – коефіцієнт дифузії парів оксиду металу в оксиді вуглецю.
– коефіцієнт дифузії парів оксиду металу в оксиді вуглецю.
Аналіз виразів (4.7)–(4.9) дозволяє стверджувати, що першою умовою підвищення швидкості випаровування оксиду і швидкості відновлення є низькі значення Х, що досягаються під час відновлення шихти, що складається з дисперсних частинок вуглецю і оксиду. При цьому, чим вища дисперсність частинок, тим менше значення Х.
Варто відзначити, що на початковій стадії відновлення оксид, що випарувався, зразу вступає в контакт з вуглецем і відновлюється, тобто
Р 2 = 0. Надалі на поверхні частинок вуглецю утворюється шар проміжного продукту відновлення чи металу, і на процес відновлення, поряд з випаровуванням оксиду і його надходженням до поверхні вуглецю, буде впливати процес доставляння оксиду до реакційної зони через цей шар і продуктів відновлення у зворотному напрямі. Якщо це застосовувати до виразу (4.9), Р 2 і Х будуть із часом зростати, і загалом швидкість процесу відновлення буде уповільнюватись.
Ще одна умова прискорення процесу відновлення – забезпечення відведення оксиду вуглецю із зони реакції і зниження його тиску (Р СО), що досягається збільшенням газопроникності шихти через зменшення дисперсності, – вступає у суперечність з першою і не завжди прийнятна.
Отже, в разі відновлення вуглецем, як і у випадку застосування водню чи його суміші з іншими газами, важливого значення набуває газопроникність відновлюваної шихти. Для цього доцільно використовувати для комбінованого відновлення гранульовану чи у вигляді брикетів шихту, що складається з вуглецевмісної сировини та оксидів металів, а як газоподібний відновник – конвертований газ. Такий відновлювальний процес набув поширення у технологічних процесах виробництва залізного порошку на Дніпровському м алюмінієвому заводі, НПО «Тулачермет», а раніше – на Броварському заводі порошкової металургії. Меха-нізм відновлення такий: дрібні частинки вуглецевої сировини рівномірно розміщуються в порах гранульованої чи збрикетованої шихти, а СО2 і Н2О, що утворюються під час відновлення, взаємодіють у порах шихти з вуглецем і регенерують СО і Н2, які знову вступають у процес відновлення. У міру витрачання вуглецю на регенерацію СО і Н2 пористість шихти не тільки зберігається, але й може збільшуватись, що створює сприятливі умови для додаткового проникнення газів-відновників з робочого простору печі в глибину відновлюваних брикетів. Такий механізм комбінованого відновлення дозволяє зберегти високу швидкість відновлення на всіх етапах процесу, що забезпечує високу продуктивність методу і якість отриманої продукції. Залізний порошок, отриманий таким чином, містить менше 0,1% вуглецю. Майже повністю виключається можливість насичення вуглецем продукту за рахунок участі на кінцевих стадіях відновлення високоактивного відновлювального газу з переважним умістом водню, а також високої газопроникності шихти.
|
|
|
|
|
Дата добавления: 2014-11-20; Просмотров: 581; Нарушение авторских прав?; Мы поможем в написании вашей работы!