КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Методи добування
|
|
|
|
1. Дегідрування бутан-бутиленової бо пентан-пентенової фракції:
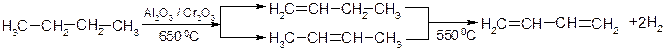
2. Дегідратація 1,3- та 1,4- гліколей:
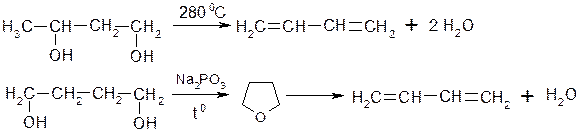
3. Промисловий метод Лєбєдєва із спирту:
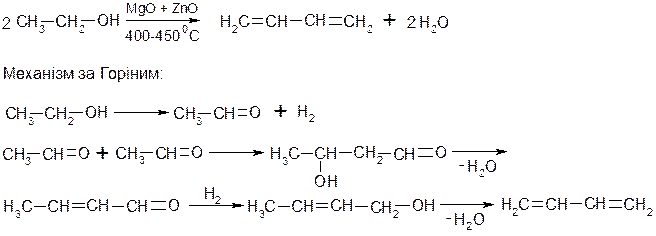
ЕЛЕКТРОННА БУВОВА СПРЯЖЕНИХ ДІЄНІВ
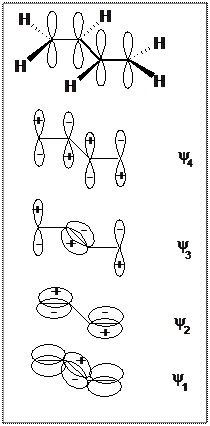 Чотири атоми карбону в молекулі бутадієну знаходяться в –гібридизованому стані і лінійно сполучені трьома σ- зв’язками, які спрямовані в просторі під кутами 1200 і утворюють остов плоскої молекули. Атоми карбону утворюють також шість σ-зв’язків з атомами гідрогену. Після утворення
Чотири атоми карбону в молекулі бутадієну знаходяться в –гібридизованому стані і лінійно сполучені трьома σ- зв’язками, які спрямовані в просторі під кутами 1200 і утворюють остов плоскої молекули. Атоми карбону утворюють також шість σ-зв’язків з атомами гідрогену. Після утворення
σ-зв’язків у молекулі бутадієну-1,3 залишаються ще чотири
р-орбіталі (по одному негібридизованому р-електрону в кожному атомі карбону), хмари яких перпендикулярні до площини молекули. При бічному перекриванні електронна густина цих чотирьох р-орбіталей зливається, утворюючи чотири МО, енергія яких поступово зростає: дві зв’язуючи π-МО та дві розпушуючи π-МО (ψ3 та ψ4):
Зв’язуючи π-Моψ1 з мінімальною енергією відповідає бутадієну-1,3 з делокалізованими π-зв’язками, що приводить до утворення єдиної π-МО, яка охоплює всі чотири атоми карбону і є спільною для всієї молекули. Утворення спільної π-МО в молекулі бутадієну-1,3 відбувається внаслідок того, що між собою перекриваються не тільки р-АО атомів С1 і С2, С3 і С4, а також р-АО атомів С2 і С3. Отже, електронні хмари двох
π-зв’язків у молекулі бутадієну взаємодіють між собою. Така взаємодія називається π,π-спряженням.
Зв’язуюча π-МОψ2 з дещо більшою енергією відповідає бутадієну-1,3 з ізольованими π-МО подвійних зв’язків.
Відповідно, бутадієн-1,3 можна розглядати як резонансний гібрид структур І – ІV:
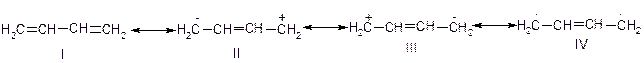
Структури ІІ – ІV вказують на те, що зв’язок між атомами С2 і С3 частково подвійний. Структура ІV зображує бірадикальний характер молекули.
Спряжені дієни відрізняються від простих алкенів тим, що вони:
а) Мають підвищену енергію утворення. Теоретично обчислена енергія утворення бутадієну-1,3 повинна становити 3269,3 кДж/моль, експериментально знайдена енергія утворення 3294,3 кДж/моль. Різниця, яка в даному випадку дорівнює 15 кДж/моль, називається енергією спряження або енергією резонансу. Отже реальна молекула бутадієну приблизно на 15 кДж/моль стійкіша, має менший запас енергії. Енергія спряження – це енергія відносної стабілізації спряжених дієнів порівняно з ізольованими дієнами. Експериментально енергію спряження можна визначити при порівнянні значень теплот гідрування спряжених та не спряжених дієнів:
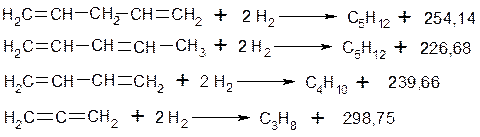
Внаслідок спряження змінюються довжини зв’язків С-С:
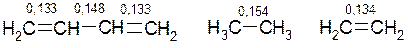 і порядок зв’язків С-С:
і порядок зв’язків С-С: 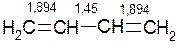 Отже, в молекулі бутадієну-1,3 спостерігається вирівнювання віддалей між атомами карбону.
Отже, в молекулі бутадієну-1,3 спостерігається вирівнювання віддалей між атомами карбону.
б) Більш реакційно здатні, внаслідок більш легкої поляризації, більшої
π-електронної густини та більшої стабілізації перехідного стану.
в) Вступають в реакції 1,2- та 1,4 приєднання.
ХІМІЧНІ ВЛАСТИВОСТІ
Для спряжених дієнів, як і для алкенів, характерні реакції приєднання.
1. Гідрування.
а) Атомарний водень приєднується переважно в положення 1,4:
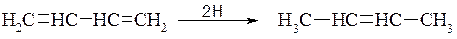
б) Водень при наявності каталізатора приєднується в положення 1,2- і 1,4:
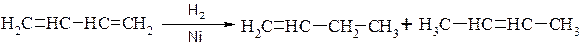
2. Приєднання галогенів
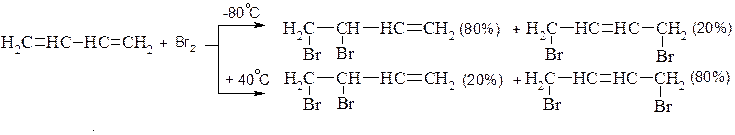 Механізм:
Механізм: 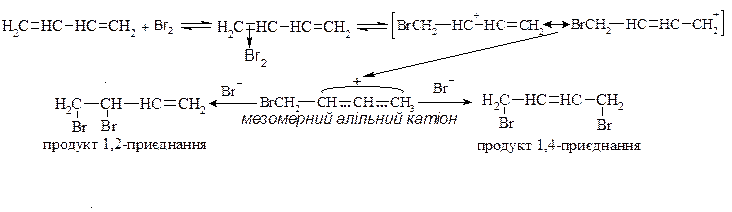
3. Приєднання галогеноводнів. Відбувається аналогічно приєднанню галогенів.
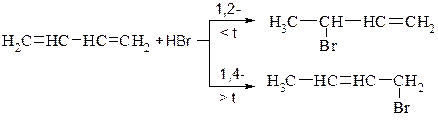
4. Гіпогалогенування – незалежно від умов реакції приводить переважно до продукту 1,2- приєднання, так як цей продукт і швидше утворюється, і більш термодинамічно стійкіший.
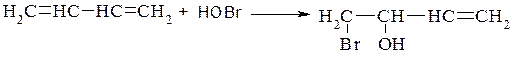
5. Дієновий синтез. Реакція Дільса-Альдера. Дієновий синтез називають реакцію 1,4-приєднання, з утворенням ненасичених шестичленних циклічних сполук:
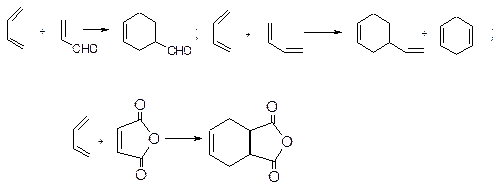
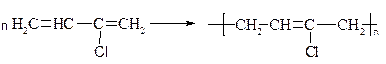
6. Полімеризація.
Природний каучук: цис-1,4-поліізопрен. Гутаперча: транс-1,4-поліізопрен
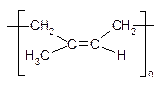
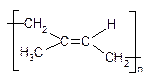
а) Синтетичний каучук СКБ (каталізатори Ціглера-Натта - AlEt3 + TiCl4 ):
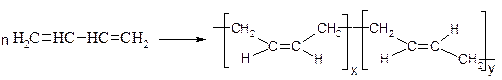
б) СКС:
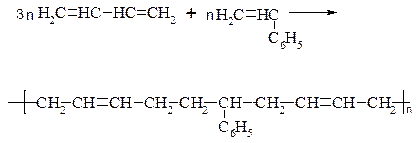
в) СКН:
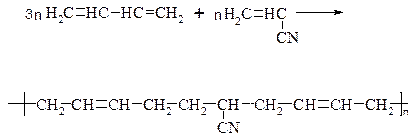
г) СКХ:
АЛКІНИ (АЦЕТИЛЕНОВІ ВУГЛЕВОДНІ)
Алкінами називають ненасичені вуглеводні, молекули яких містять між карбоновими атомами потрійний зв’язок 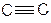 . Загальна формула CnH2n-2.
. Загальна формула CnH2n-2.
МЕТОДИ ДОБУВАННЯ АЛКІНІВ
І. Промислові.
1. Окислювальний піроліз алканів:
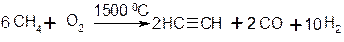
2. Піроліз алканів:
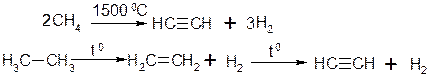
3. Карбідний метод:
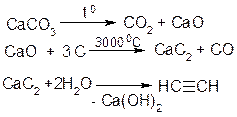
ІІ. Лабораторні:
1. Дегідрогалогенування віцинальних та гемінальних дигалогеналканів:
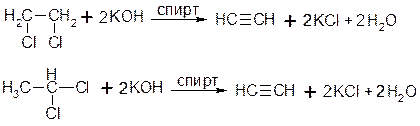
2. Алкілування ацетиленідів
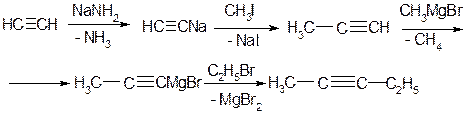
БУДОВА
Потрійний зв’язок в ацетилені являє собою комбінацію σ- зв’язку, який утворюється внаслідок перекривання двох sp-гібридних орбіталей атомів карбону та двох π- зв’язків, які утворюються внаслідок попарного перекривання чотирьох р-орбіталей.

1. Довжина зв’язку в ацетилені становить 0,121 нм, тобто коротша довжини подвійного зв’язку в етилені (0,134 нм); в етані (0,154 нм). Скорочення зв’язку обумовлено:
а) участю в утворенні σ- зв’язку двох sp-гібридних орбіталей з меншими власним радіусом;
б) допоміжним скороченням зв’язку за рахунок утворення двох π- зв’язків.
2. Енергія зв’язку в ацетилені набагато більша, ніж в етилені чи етані. Загальна енергія зв’язку 812 кДж/моль. Але якщо енергія зв’язку С-С в етані становить 344 кДж/моль то в середньому енергія π- зв’язку становить лише 234 кДж/моль, тобто на 110 кДж/моль менша енергії σ- зв’язку. Відповідно, для ацетилену будуть характерні реакції з розривом зв’язку . Але при цьому...
3. Енергія іонізації зв’язку (енергія, необхідна для вибивання електрону з π- зв’язку) дорівнює 11,4 еВ, що значно більше ЕІ СН2=СН2 – 10,5 еВ та практично дорівнює ЕІ СН3–СН3 – 11,6 еВ. Отже, електрони π- зв’язків в ацетилені міцно утримуються атомами карбону і вони значно менш придатні для атаки електрофілом, ніж π-електрони алкенів. Це відповідає sp-гібридизації атому карбону. При цьому енергія іонізації ацетиленових вуглеводнів різко зменшується при введенні алкільних груп. Так для пропілену ЕІ становить лише 10,4 еВ. Отже, пропілен в реакціях електрофільного приєднання буде реагувати значно легше, ніж ацетилен. Питання чому? Тому, що потрійний зв’язок легко поляризується і заміщені ацетилени більш поляризовані, ніж алкени. Наприклад, дипольний момент пропілену становить 0,35D, тоді як пропіну – 0,75D.
4. Довжина зв’язку С-Н в ацетилені також менша, ніж в алкенах і алканах і відповідно становить 0,106, 0,108, 0,109 нм. Відповідно, енергія зв’язку С-Н в ацетилені більша. Тобто, sp-гібридизація атому карбону в ацетилені зашкоджує гомолітичному розриву зв’язку С-Н з утворенням вільних радикалів. Але внаслідок більшої поляризації зв’язку С-Н в ацетилені із-за більшої електронегативності sp-гібридизованого атому карбону (2,75, 2,62 та 2,50 для sp, sp2, sp3 - гібридизованих атомів карбону) полегшується гетеролітичний розрив зв’язку С-Н з утворенням йонів. КдисС2Н2 = 10-22, тоді як КдисС2Н6 = 10-40.
ХІМІЧНІ ВЛАСТИВОСТІ
Алкіни вступають в реакції приєднання, які можуть відбуватися за радикальним, електрофільним та нуклеофільним механізмами, а також в реакції заміщення атомів водню, розміщених біля атому карбону з потрійним зв’язком.
І. Приєднання радикальних реагентів. Гідрування. Потрійний зв’язок гідрується легше, ніж подвійний зв’язок в алкенах.
а) Водень в момент народження:
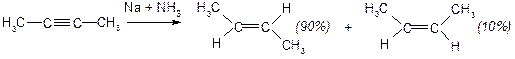
б) Каталітичне гідрування:
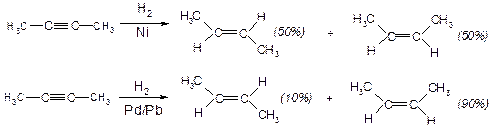
Реакції, які переважно приводять до одного з кількох можливих стереоізомерів називають стереоселективними; реакції, які приводять до одного з кількох можливих – стереоспецифічними.
ІІ. Приєднання електрофільних реагентів.
1. Галогенування – може відбуватися, як за радикальним, так і електрофільним механізмом. Електрофільне галогенування відбувається транс-стереоселективно:
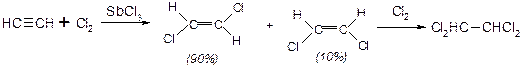
2. Гідргалогенування – відбувається також транс-стереоселективно, за правилом Марковнікова.
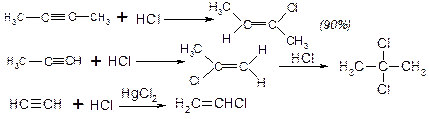
ІІІ. Приєднання нуклеофільних реагентів.
1. В присутності каталізаторів, які збільшують електрофільність потрійного зв’язку. Приєднання Н2О.
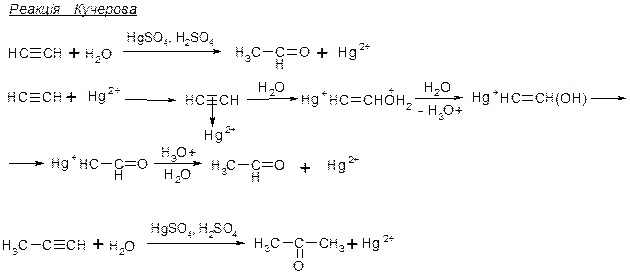
Правило Ельтекова: ненасичені спирти з групою ОН біля атому карбону з подвійним зв’язком нестійкі і самовільно ізомеризуються в стійкі карбонільні сполуки. Між енольною та карбонільною сполуками існує рівновага:
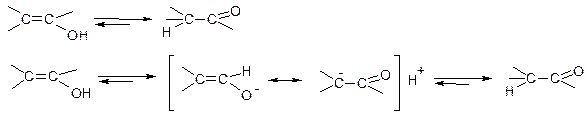
Причина ізомеризації – висока полярність зв’язку ОН. При приєднанні протону знову до атому оксигену, він може знову відщеплюватися, при приєднанні до атому карбону – значно гірше, тобто поляризація зв’язку С-Н значно менша. В результаті відбувається перегрупування, при якому сильніша кислота перетворюється на слабшу. Структурні ізомери, між якими в нормальному стані існує рівновага називаються таутомерами.
2. В присутності каталізаторів, які збільшують нуклеофільність реагентів.
a. Приєднання спиртів з утворенням вінілових ефірів:
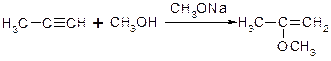
b. Приєднання синільної кислоти з утворенням нітрилів:
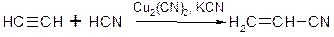
c. Приєднання оцтової кислоти:
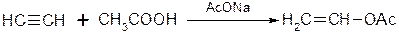
ІV. Карбонолування алкінів
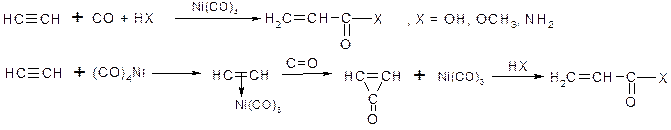 V. Реакції заміщення:
V. Реакції заміщення:
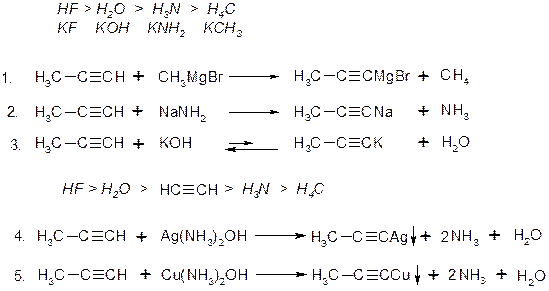
VІ. Реакції приєднання до альдегідів та кетонів:
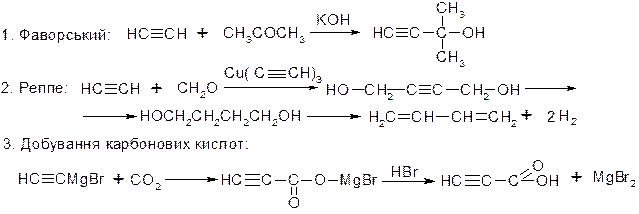
VІІ. Реакції заміщення атому гідрогену біля потрійного зв’язку на галоген:

VІІІ. Реакції полімеризації:
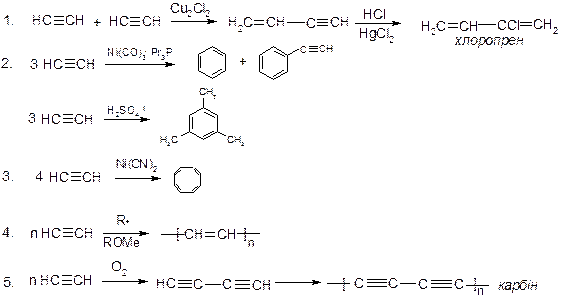
ІХ. Реакції окиснення:
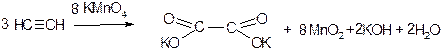
АЛІЦИКЛІЧНІ СПОЛУКИ
Аліциклічні сполуки – це аліфатичні і циклічні вуглеводні, кільце (цикли) яких побудовані тільки з вуглецевих атомів. Назви аліциклічних сполук утворюють шляхом додавання префіксу цикло - до назви відповідного насиченого вуглеводню. Наряду з циклопарафінами існують циклоолефіни тощо.
Для зручності аліциклічні сполуки часто зображують у вигляді простих геометричних фігур: циклопропан – трикутником, циклобутан – квадратом тощо. При цьому розуміється, що у кожного з кутів фігури знаходиться по два атоми гідрогену, якщо не визначена наявність інших груп.
Для аліциклічних сполук характерна структурна ізомерія таких видів:
1. За числом атомів карбону в циклі:
2. За числом атомів карбону в замісниках:
3. За розміщенням замісників у циклі:
4. Геометрична ізомерія:
5. Оптична ізомерія при відсутності площини симетрії:
НАПРУЖЕННЯ У ЦИКЛАХ. Моноциклопарафіни залежно від розміру циклу поділяють на циклоалкани з малим (3-4), нормальним (5-7), середнім
(8-11) і великим (макро) (12 і більше) циклом. При цьому в залежності від величини циклу циклоалкани мають різні стійкість. Малі цикли нестійкі і в хімічних реакціях та при нагріванні легко розриваються, тоді як інші цикли мають високу міцність.
Для пояснення цього явища Байєр у 1885 році запропонував теорію напруження, згідно з якою мірою стійкості циклу є відхилення валентних кутів у циклах від нормального тетраедричного значення 109028΄. Чим більше відхилення, тим меншу міцність має цикл. Наприклад:
Мірою напруження циклів за Байєром є половина різниці між тетраедричним кутом і валентним кутом атому карбону в циклі при розміщенні всіх атомів карбону в одній і тій же площині. Теорія Байєра змогла пояснити, чому відомі вже на той час три та чотиричленні цикли нестійкі і чому цикл у молекулі циклопентану стійкіший. Проте з цієї гіпотези виходило, що вже циклогексан і інші багаточленні цикли повинні мати напруження і це напруження повинно зростати з розміром циклу. В дійсності ж цього не відбувається. Так, про напруження в циклі можна судити за теплотою згоряння циклічних сполук: чим більша теплота виділяється при згорянні на одну метиленову групу СН2, тим більше напруження в циклі. Експериментально встановлено, що теплота згоряння, яка приходиться на одну групу СН2, найменша для циклогексану. Якщо напруження в цьому циклі прийняти за “0”, то напруження трьохчленному циклі становить 38,5 кДж/моль тощо, а в циклах з числом атомів більше шести – в межах 0-6 кДж/моль. Тобто, з аналізу теплот згоряння виходить, що лише малі цикли мають напруження. Що ж невірно в теорії Байєра?
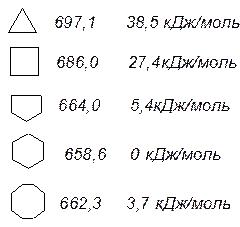
Згідно з сучасними уявленнями, атоми карбону в циклі окрім циклопропанового, розміщується в просторі не в одній площині. Зокрема, для циклогексану були запропоновані конформації 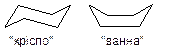 , що дозволяє наблизити величини валентних кутів атомів карбону до значення 109028΄.
, що дозволяє наблизити величини валентних кутів атомів карбону до значення 109028΄.
Сучасна теорія енергетичного стану молекул, а отже і їх стійкості, враховує не лише стан валентних кутів, але й можливість обертання навколо зв’язків і взаємне відштовхування атомів в молекулах. Це дало можливість сформулювати загальну теорію напруження в циклах.
1. У циклах існують різні види напруження.
2. Кутове (байєрівське) напруження виникає в результаті відхилення валентних кутів від кута 109028΄.
3. Конформаційне (пітцеровське) напруження виникає в результаті вимушеного повороту зв’язків і відхилення від енергетично найвигідніших загальмованих конформацій. При цьому виникають крутильні (торсійні) сили, які приводять до інверсії циклу – взаємного перетворення конформаційних форм.
4. Стеричне (Ван-дер-Ваальсівське) напруження виникає внаслідок взаємного відштовхування зближених у просторі атомів гідрогену та груп замісників, яке має місце у випадках, коли атоми або групи атомів зближаються на відстань, меншу за суму їх ван-дер-Ваальсових радіусів.
5. Диполь-диполне напруження обумовлене відштовхуванням однойменних диполів. Атоми або групи атомів завжди намагаються зайняти таке положення, в якому спостерігається мінімальне диполь-дипольне відштовхування та максимальне диполь-диполне притяжіння.
6. Напруження в циклі може виникати також як результат зміни міжатомних відстаней за рахунок збільшення або зменшення довжини зв’язків.
В малих циклах існують всі види напруження.
В нормальних циклах відсутнє кутове напруження, а в конформації “крісло” циклогексану відсутні будь-які напруження. Конформація “крісло” на 28,5 кДж/моль вигідніша, ніж конформація “ванна”, в якій є конформаціне та стеричне напруження. В конформаціях циклогексану та інших циклоалканів виділяють два типи зв’язків С-Н - та екваторіальні. В циклогексані відбувається швидкий перехід з однієї конформації “крісло” в іншу, тобто йде процес інверсії циклу. Замісник в циклогексані завжди намагається зайняти екваторіальне положення як енергетично більш вигідне, внаслідок стеричного напруження.
Макроцикли не мають кутового, конфірмаційного та стеричного напружень.
МЕТОДИ ДОБУВАННЯ
І. Загальні методи.
1. Дегалогенування α, ω-дігілогенпохідних алканів металічним цинком або натрієм.
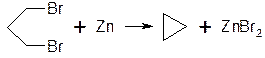
2. Взаємодія дігалогенпохідних алканів з натріймалоновим ефіром.
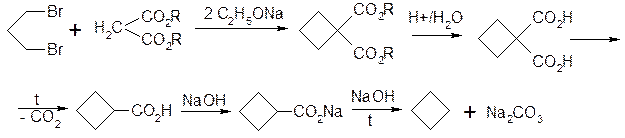
3. Відщеплення галогеноводнів від галоген карбонільних сполук.
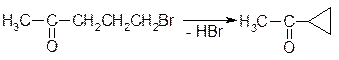
4. Піроліз солей дикарбонових кислот та самих кислот, придатних до добування циклічних сполук з п’яти та більше атомами карбону в циклі.
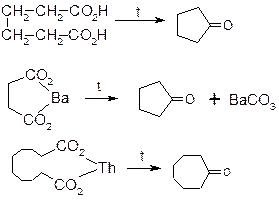
ІІ. Спеціальні методи.
1. Дієновий синтез: 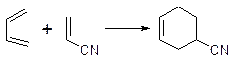
2. Гідрування ароматичних вуглеводнів: 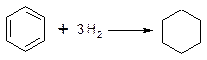
Електронна будова циклопропану. В дійсності валентні кути між атомами карбону в циклопропані становлять не 600, як у правильного трикутнику, але й не 109028΄, як у алканах, а дорівнюють 1040. Такий валентний кут досягається проміжним типом перекривання гібридизованих орбіталей атомів карбону між осьовим (σ) та боковим (π) перекриванням. В дійсності реалізується “банановий” зв’язок, проміжний між σ- та π- зв’язками, оскільки в такому відбувається деяка де локалізація електронів за типом кільцевої π-електронної хмари, яка оточує цикл і розміщена в одній з ним площині. Оскільки “банановий” зв’язок проміжний, він здатний вступати до реакцій приєднання, як ненасичені сполуки (етилен), але значно гірше. В той же час валентний кут Н-С-Н в циклопропані становить 1180 і майже дорівнює валентному куту в етилені. Звісно, і гібридизація атомів карбону проміжна між sp3 та sp2.
ХІМІЧНІ ВЛАСТИВОСТІ
Хімічні властивості циклоалканів визначаються в основному розміром циклу. Для 3-х і 4-х членних циклів характерні реакції приєднання, які супроводжуються розривом циклу, тоді як для всіх інших циклоалканів – характерні реакції заміщення, як і для алканів.
1. Гідрування:
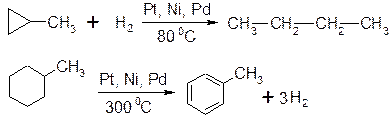
2. Галогенування:
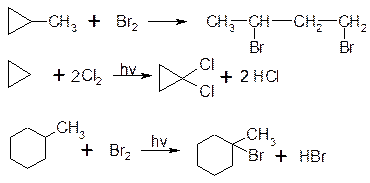
3. Гідрогалогенування:
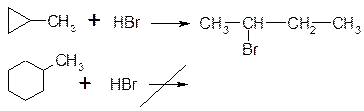
4. Взаємодія з сильними кисеньвмісними кислотами:
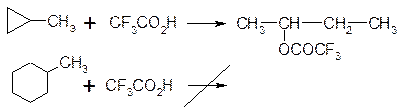
5. Окиснення. Циклоалкани не реагують з озоном і з розведеними розчином KMnO4. Під дією сильніших окислювачів (нітратної кислоти, кисень при наявності каталізаторів, концентрований розчин KMnO4) відбувається поступове окиснення з розривом циклу і утворенням дикарбонових кислот:
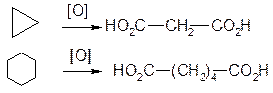
6. Реакція розширення циклу:
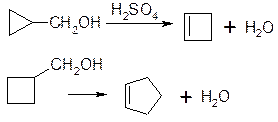
7. Реакція звуження циклу:
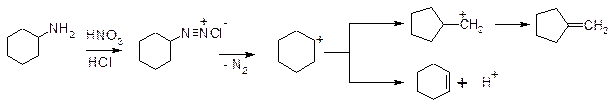
АРОМАТИЧНІ ВУГЛЕВОДНІ
До ароматичних сполук відносяться бензол та інші сполуки, які нагадують його за хімічними властивостями. Бензол має особливі властивості, які відрізняють його від аліфатичних вуглеводнів: аліфатичні ненасичені вуглеводні головним чином вступають в реакції приєднання, аліфатичні насичені вуглеводні – в реакції вільно-радикального заміщення, а ароматичні вуглеводні – головним чином в реакції йонного заміщення.
Основний представник ароматичних сполук – бензол – вперше виділив у 1825 році Фарадей із світильного газу. Його хімічні властивості вивчені краще, ніж будь-якої іншої органічної сполуки. Незважаючи на це, лише у 1931 році була запропонована задовільна структура для бензолу, яка згодом стала загальноприйнятою.
У 70-х роках ІХ століття було вже відомо, що бензол має молекулярну формулу С6Н6. у 1865 році Кекуле запропонував зображати структурну формулу бензолу у вигляді 6-членного циклу з трьома подвійними зв’язками. Але ця формула не пояснювала основні хімічні властивості ароматичних сполук, завдяки яким вони і були виділені в особливий клас. Ці властивості і становлять хімічні критерії ароматичності:
1. Легкість утворення ароматичних сполук в різноманітних реакціях:
2. Стійкість до дії слабких окиснювачів на відміну від ненасичених сполук:
3. Інертність до реакцій електрофільного приєднання:
4. Легкість реакції електрофільного заміщення:
5. Атоми і групи атомів, які сполучені з бензольним ядром набувають особливих властивостей: феноли значно більш сильні кислоти, ніж спирти, а анілін слабкіша основа, ніж аліфатичні аміни.
Ця формула не пояснювала також, чому немає ізомерів у 1,2-дизаміщенних похідних бензолу, наприклад 
Тому у 1872 р. Кекуле запропонував осциляційну гіпотезу, згідно з якою подвійні карбон-карбонові зв’язки в бензольному ядрі не закріплені, а безперевно переміщуються (осцилюють) у кільці молекули. Ця гіпотеза дозволила пояснити, чому немає двох ізомерів о-ксилолу, що було доказано раніше його озонолізом.
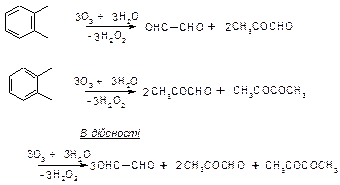
Але осциляційна гіпотеза не пояснювала інших хімічних аномалій бензолу. Сучасні дані також не підтверджують осциляційну гіпотезу.
Фізичні критерії ароматичності. Вирішальне значення для становлення структури бензолу мали фізичні методи дослідження. Було встановлено, що:
1. Молекула бензолу являє собою плоский правильний шестикутник з валентним кутом 1200. Довжина всіх карбон-карбонових зв’язків однакові і дорівнюють 0,14 нм і мають приблизно середнє значення між довжинами простих (0,154 нм) т подвійних (0,134 нм) карбонових зв’язків. Порядок зв’язків С-С в бензолі 1,67. Довжини всіх зв’язків С-Н також однакові і становлять 0,108 нм.
2. Молекула бензолу симетрична і має вісь шостого, а не третього порядку, як випливає з формули Кекуле
3. Бензол є неполярною сполукою. Отже, ядра атомів та валентні електрони в молекулі бензолу розміщені симетрично.
4. Теплота гідрування бензолу значно менша, ніж слід було очікувати при гідруванні структури Кекуле. Так, теплота гідрування циклогексену 119,7 кДж/моль. Відповідно, при гідруванні структури Кекуле повинно було виділитись в три рази більше теплоти 359,1 кДж/моль. В дійсності виділяється лише 208,4 кДж/моль, тобто на 150,7 кДж/моль менше. Відповідно, енергія бензолу менша від енергії циклогексатрієну – 1,3,5 на цю ж величину. Тобто, бензол стабільніший від циклогексатрієну на 150,7 кДж/моль. Отже, бензол є термодінамічно стійкою сполукою і утворення його відбувається енергетично вигідно. Різниця між теоретичною енергією циклогексатрієнуж-1,3,5 та енергією бензолу називається енергією стабілізації, енергією спряження або енергією резонансу бензолу. Взагалі основними фізичними критеріями ароматичного характеру органічних сполук є їх підвищена енергетична стійкість, тобто зменшення енергії при утворенні циклічної системи із аліциклічної спряженої системи з тим же числом атомів карбону та π-електронів. Якщо при переході від ациклічної спряженої системи до циклічної з тим же числом π-електронів енергія системи не змінюється, цикл є неароматичним, якщо ж енергія системи підвищується, то цикл є анти ароматичним.
Але як а priori без фізичних досліджень можна визначити, який характер має та чи інша сполука – ароматичний, неароматичний чи антиароматичний?
Для цього існує правило кола. Якщо циклічну систему, що складається з k-атомів, які не знаходяться у sp3-гібридизації, вписати у вигляді k-кутника в коло таким чином, щоб одна із вершин знаходилась в нижній точці, тоді число вершин k-кутника відповідає числу π-МО системи, а проекція цих вершин на вісь ординат – відносним енергіям цих π-МО. При цьому π-МО, розташовані нижче центру кола, є π- зв’язуючими, розташовані на рівні центру кола - π- незв’язуючими, вище центру кола - π- розпушуючими. Якщо в циклічній системі, в якій жоден атом не знаходиться у sp3-гібридизації, зайняті π-електронами лише зв’язуючи π-МО, то ця система повинна бути ароматичною. Якщо ж зайнятими є не лише π- зв’язуючи, а й π-незв’язуючи, а тим більше π-розпушуючи МО, то система повинна бути антиароматичною.
Квантово-хімічні критерії ароматичності. Правило Хюккеля. Ароматичні властивості має будь-яка плоска моно циклічна сполука, в якій всі атоми знаходяться у sp2-гібридизації, а цикл містить 4n+2 π-електронів, де n – будь-яке число натурального ряду – 0, 1, 2, 3 тощо. Якщо така система має 4n π-електронів, вона анти ароматична, якщо ж система не плоска, то незалежно від кількості π-електронів вона не ароматична.
Бензол є типовим прикладом ароматичної системи з n=1, тобто має секстет π-електронів. Розглянемо цю систему пильніше.

В молекулі бензолу всі атоми карбону знаходяться у sp2-гібридизованому стані і при цьому дві sp2-гібридизовані орбіталі витрачаються на утворення двох σ- зв’язків С-С, одна sp2-гібридизована орбіталь – на утворення σ- зв’язку С-Н. Шість р-орбіталей, в результаті взаємодії між собою утворюють шість π-МО: три зв’язуючих та 3 розпущуючих. На зв’язуючих π-МО знаходяться шість π-електронів, на розпушуючих – жодного. При цьому найстійкіша за енергією зв’язуюча π-МО охоплює все бензольне ядро, в цій π-МО можливе кільцеве переміщення π-електронів. Всі атоми і зв’язки С-С таким чином рівноцінні. Розглядаючи ще дві вище розташовані π-МО, приходимо до такого ж висновку. Тобто в молекулі бензолу немає взагалі локалізованих подвійних зв’язків і молекулу краще всього зображати такою формулою 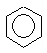 .
.
Окрім бензолу існують і інші ароматичні сполуки, які мають в циклі лише атоми карбону?
1. Аннудени:
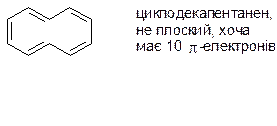
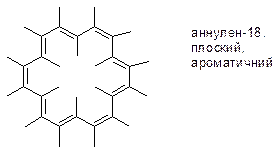
2. Карбоцикли, гомо ароматичні системи – це заряджені частини (аніони та катіони), які мають ароматичний характер, тоді як відповідні незаряджені структури мають 4n π-електронів і відповідно неароматичний характер.
a. Циклопропенілій – катіон:
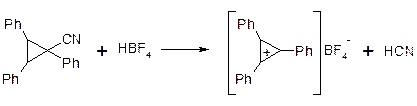
b. Циклопентадієніл-аніон:
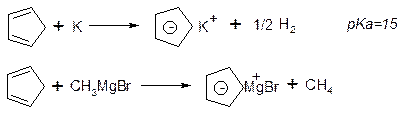
c. Ферроцен:
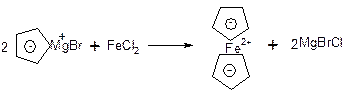
d. Тропілій –катіон:
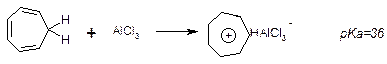
3. Гетероцикли – цикли, які містять в своєму складі інші атоми, крім карбону, також можуть бути ароматичними за рахунок включення в π-систему електронів гетеро атомів: фуран, пірол, тіофен тощо.
4. Поліциклічні ароматичні системи: нафталін, тощо.
АРОМАТИЧНІ ВУГЛЕВОДНІ РЯДУ БЕНЗОЛУ
Бензол є першим членом гомологічного ряду ароматичних вуглеводнів, склад яких відповідає формулі CnH2n-6. Гомологічна різниця, як і раніше, становить (СН2)n.
Ізомерія гомологів і похідних бензолу – зумовлена ізомерією радикалів (замісників), їх кількістю та положенням у бензольному ядрі – на приклад, етилбензол, о-, м- і п-ксилоли.
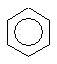
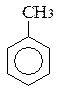
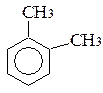
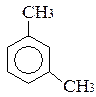
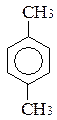
бензол толуол орто-ксилол мета-ксилол пара-ксилол
(метилбензол) (1,2-диметилбензол) (1,3-диметилбензол) (1,4-диметилбензол)
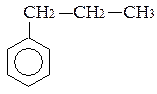
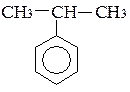
пропілбензол ізопропілбензол (кумол)
ДОБУВАННЯ БЕНЗОЛУ ТА ЙОГО ГОМОЛОГІВ
1. Основні джерела – коксування кам’яного вугілля та нафти. З метою підвищення масової частки ароматичних вуглеводнів у нафті її піддають ароматизації, в результаті чого з нафти вдається добути до 15-20% ароматичних вуглеводнів.
2. Ароматизація парафінів – Казанський, Плате, Молдавський (вміст ароматичних вуглеводнів зростає до 50%):
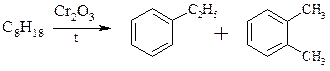
3. Синтетичні методи:
a. Реакція Вюрца-Фіттіга, механізм якої аналогічний механізму реакції Вюрца
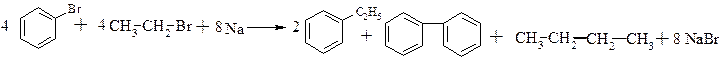
b. Алкілування ароматичних вуглеводнів. Реакція Фріделя-Крафтса (SE)
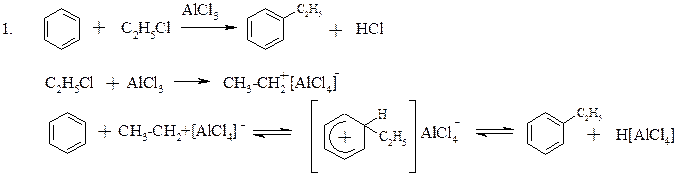
2. Реакція супроводжується ізомеризацією:
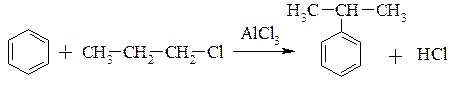
3. Реакція алкілування алкенами – видозмінена реація Фріделя-Крафтса:
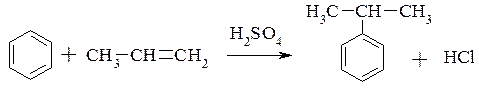
ХІМІЧНІ ВЛАСТИВОСТІ
1. Реакції приєднання:
2.
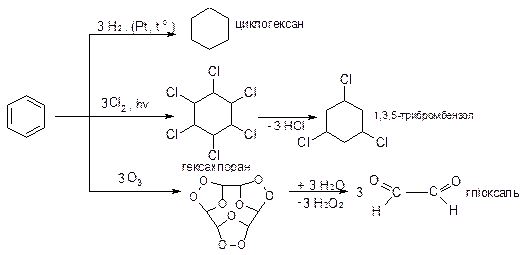 |
Реакції окислення:
а) 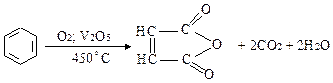
б) 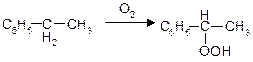
в) 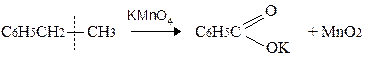
3. Реакції електрофільного заміщення:
а) Алкілування за Фріделем-Крафтсом:
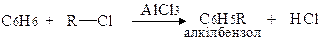
б) Ацилювання за Фріделем-Крафтсом:
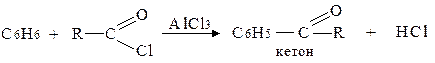
в) Галогенування
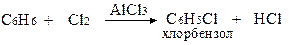
г) Сульфування
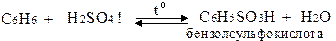
д) Нітрування
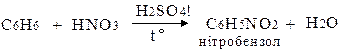
Механізм электрофільного заміщення у ароматичному ядрі (SЕ):
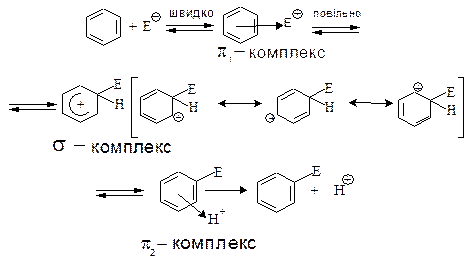
Gершою стадією реакції електрофільного заміщення є утворення π-комплексу – перехідного стану, який виникає із ароматичної сполуки та електрофільного реагенту за рахунок електростатичних сил притягнення між позитивно-зарядженим електрофільм та π-електронами ароматичного сполуки. Утворення π-комплексу проходить дуже швидко, енергія активації утворення π-комплексу (енергія, необхідна для утворення π--комплексу) складає приблизно 35 кДж/моль.
Другою стадією реакції, визначаючої її швидкість, є перехід π-комплексу у σ-комплекс. σ- Комплекс – це проміжна сполука, яку в окремих випадках можна зареєструвати або навіть виділити і індивідуальному стані, як, наприклад, 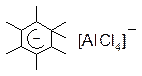 . σ-Комплекс – це катіон, який вже не має ароматичних властивостей. В процесі утворення σ-комплексу атом карбону, який приєднав електрофіл, переходить із тригонального sp2-гібридизованого стану в тетраедричний sp3-гібридизований стан. При цьому між атомом карбону ароматичного ядра та електрофілом виникає σ- зв’язок за рахунок пари електронів ароматичного секстету. У бензольному ядрі внаслідок цього залишається тільки чотири π-електрони і воно набуває позитивного заряду. Чотири π-електрони делокалізовани на п’яти атомах sp2-гібридизованого карбону. У σ- комплексі як частина Х, так і атом гідрогену при карбоні, який несе замісник Х, розміщені у вершинах тетраедру і не знаходяться в одній площині з бензольним ядром.
. σ-Комплекс – це катіон, який вже не має ароматичних властивостей. В процесі утворення σ-комплексу атом карбону, який приєднав електрофіл, переходить із тригонального sp2-гібридизованого стану в тетраедричний sp3-гібридизований стан. При цьому між атомом карбону ароматичного ядра та електрофілом виникає σ- зв’язок за рахунок пари електронів ароматичного секстету. У бензольному ядрі внаслідок цього залишається тільки чотири π-електрони і воно набуває позитивного заряду. Чотири π-електрони делокалізовани на п’яти атомах sp2-гібридизованого карбону. У σ- комплексі як частина Х, так і атом гідрогену при карбоні, який несе замісник Х, розміщені у вершинах тетраедру і не знаходяться в одній площині з бензольним ядром.
Утворення σ-комплексу потребує значно більше енергії, ніж утворення π-комплексу. Але, в цілому, утворення σ-комплексу не потребує надто великої затрати енергії, оскільки енергія спряження σ-комплексу (109 кДж/моль) близька до енергії бензолу (150 кДж/моль).
 |
пари Рис. Енергетична діаграма реакції SE для бензолу
Е1 – енергія бензолу; Е2 – енергія активації утворення π1 – комплексу; Е3– енергія π1 – комплексу; Е4 – енергія активації утворення s - комплексу; Е5– енергія s - комплексу;
Е6 – енергія активації утворення π2 – комплексу; Е7– енергія π2 – комплексу; Е8– енергія активації утворення продукту реакції; Е9– енергія продукту реакції.
Третя стадії реакції – утворення нового π-комплексу за рахунок повернення електронів, яка спонукала sp3-гібридизований стан атому карбону з атомом гідрогену, в бензольний цикл і утворення таким чином знову ароматичного секстету.
Зміна вільної енергії системи реакції електрофільного заміщення зображується такою кривою:
ПРAВИЛА ОРІЄНТАЦІЇ ЕЛЕКТРОФІЛЬНОГО ЗАМІІЩЕННЯ В БЕНЗОЛЬНОМУ ЯДРІ
В молекулі бензолу всі атоми карбону рівноцінні, тому електрофільне заміщення може з однаковою імовірністю проходити по любому з них. Якщо ж в бензольному ядрі є замісники, то реакційна здібність атомів карбону буде залежати від таких факторів:
1. Положення та природа замісників;
2. Природа реагенту;
3. Умови проведення реакцій.
Замісники за впливом на орієнтацію електрофільного заміщення поділяють на дві групи.
А) Замісники першого роду або о-, п- орієнтанти, які направляють новий замісник в орто- і пара- положення по відношенню до себе. Замісники першого роду – це атоми або групи атомів, здатні віддавати електрони (донори електронів). Як правило, всі замісники І-го роду (за винятком алкілів) мають на атомі, безпосередньо зв’язаним з ароматичним кільцем, по меншій мірі, одну неподілену пару електронів.

Б) Замісники другого роду або мета- орієнтанти направляють новий замісник в мета- положення по відношенню до себе. Замісники другого роду – це акцептори електронів. Як правило, замісники ІІ-го роду мають на атомі, безпосередньо з’єднаним з ароматичним кільцем повний або частково позитивний заряд. Частіше всього ці атоми мають подвійний зв’язок.
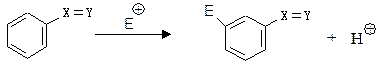
Замісники впливають не лише на орієнтацію заміщення, але і на його швидкість. За впливом на швидкість реакції електрофільного заміщення замісники І-го роду поділяють на чотири групи:
I. Сильноприскорюючи реакції SE:
II. Помірноприскорюючи реакції SE:
III. Малоприскорюючи реакції SE:
IV. Уповільнюючи реакції SE:
Замісники ІІ-го роду по цій ознаці поділяють на три групи:
I. Сильноуповільнюючи реакції SE:
II. Помірноуповільнюючи реакції SE:
III. Малоуповільнюючи реакції SE:
Замісники І-го роду (за винятком галогенів) прискорюють реакції SE у всіх положеннях бензольного кільця. Орієнтуючий вплив цих замісників обумовлений передусім тим, що о- і п-орієнтанти сильніше всього прискорюють реакції в о- і п- положеннях по відношенню до себе.
Замісники ІІ-роду, навпаки, уповільнюють реакції SE у всіх положеннях бензольного кільця. Орієнтуючий вплив мета-замісників обумовлений тим, що вони найбільш уповільнюють реакції в о- і п- положеннях, тобто найменше – в м- положеннях, по яким і відбувається реакція. Іншими словами, замісники І-го роду активують бензольне кільце за рахунок подавання електронів у кільце, тоді як мета-орієнтанти дезактивують бензольне кільце за рахунок відтягування електронів з кільця.
Механізм орієнтуючого впливу замісників визначається структурою замісників та ефектами взаємодії замісники і бензольного ядра. Відрізняють два фактори:
1. Статичний вплив замісників – розподіл електронної густини в ізольованій не реагуючий молекулі під впливом індукційного ефекту та ефекту спряження:
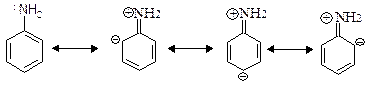
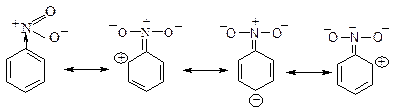
Проте вирішальне значення для орієнтації електрофільного реагенту має динамічний фактор.
2. Динамічний фактор – вплив замісників на стійкість σ-комплексу, що утворюється. Енергія активації реакції електрофільного заміщення тим менша, а швидкість реакції тим більша, чим вище стійкість проміжного σ-комплексу, чим менше енергії необхідно для його утворення.
Відповідно, якісний аналіз орієнтаційного впливу любого замісника зводиться до аналізу впливу цього замісника на стійкість σ-комплексу, який утворюється при реакції. Оскільки головним фактором, що впливає на стабільність σ-комплексу є спряження, в першу чергу повинна аналізуватися можливість максимального спряження при утворенні σ-комплексу за участю різних атомів бензольного кільця. Відповідно, стійкість σ-комплексу емпірично можна визначити таким чином – вона зростає із збільшенням числа можливих межових структур σ-комплексу, тобто збільшується із зростанням делокалізації заряду по молекулі. При цьому межові структури вносять різний внесок в стабілізацію σ-комплексу. Найбільший внесок вносять структури, в яких:
а) максимальне число π- зв’язків;
б) всі атоми (крім гідрогену) мають октетну (заповнену) оболонку;
в) заряд виноситься за межі бензольного кільця.
В той же час практично не мають значення для стабілізації σ-комплексу межові структури, в яких однойменні заряди розташовані на сусідніх атомах, і, тим більше, на одному й тому ж атомі.
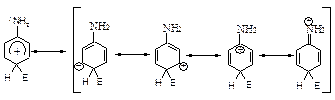
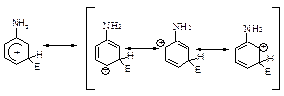
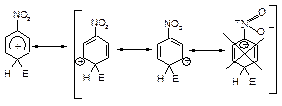

Правила написання межових структур
1. При написанні межових структур атоми ніколи не змінюють свого положення.
2. Заряд ніколи не може знаходитися на sp3-гібридному атомі карбону. Він знаходиться в бензольному кільці лише в о- і п- положеннях (сусідніх та протилежному атомах карбону) по відношенню до sp3-гібридизованого атому карбону кільця.
3. Подвійними зв’язками з’єднуються атоми карбону кільця, які не знаходяться в sp3-гібридизованому стані і які не мають заряду.
4. Якщо заряд знаходиться на атомі карбону., який несе замісник І-го роду, завжди реалізуються межові структури, в яких заряд виноситься за межі ароматичного кільця.
Правила орієнтації не мають характеру закону. Описані раніше закономірності вказують лише на головний напрямок реакцій електрофільного заміщення. Дуже часто в реакціях утворюються всі можливі ізомери. Однак, мають перевагу ті, які утворюються в результаті описаних вище закономірностей.
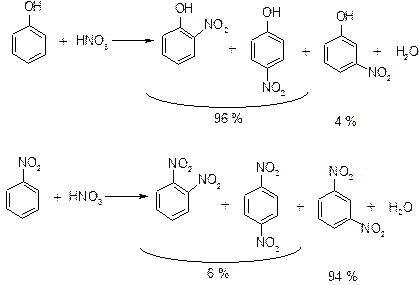
Співвідношення о- і п-ізомерів в реакціях електрофільного заміщення визначається такими факторами:
1. В бензольному ядрі є два орто-положення і одно пара. Тому о-ізомерів утворюється при інших рівних умовах в два рази більше.
2. Наявність об’ємного замісника зашкоджує входженню реагенту в орто-положення.
3. Індукційний ефект сильніше проявляється в орто-положенні, тоді як ефект супряження – в пара-положенні за рахунок утворення більш стійкої пари-хіноїдноої межової структури σ-комплексу.
При наявності в молекулі бензолу 2-х замісників їх орієнтація може бути узгодженою та неузгодженою. Узгоджена орієнтація – це коли обидві замісники направляють електрофільний реагент в одні і тій ж положення. Неузгоджена орієнтація – навпаки.
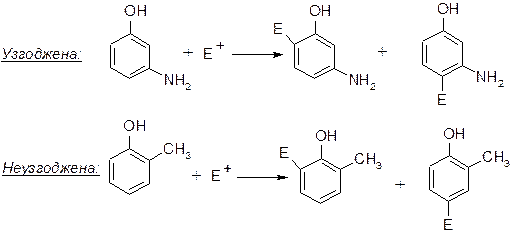
При неузгодженій орієнтації замісників місце входження електрофільного реагенту визначається такими правилами:
1. Вплив замісника, сильноприскорюючого реакції SE завжди переважає вплив інших замісників І-го роду і тим більше замісників ІІ-го роду. Це явище обумовлено утворенням більш стійкого σ-комплексу.
2. Як правило, електрофільний реагент ніколи не входить в положення, яке знаходиться між мета-розташованими замісниками. Це обумовлено просторовими утрудненнями входження нового замісника.
ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ
- циклічні системи в молекулах яких крім атома вуглецю знаходиться гетеро атом (О, N, S). Класифікуються на п’ятичленні цикли, шестичленні цикли. Гетероциклічні сполуки поширені у природі – вони входять до складу хлорофілу і гемоглобіну, багатьох біологічно активних сполук (медикаментів), барвників тощо.
ПЯТИЧЛЕННІ ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ
Ароматичність п’ятичленних гетероциклів – фурану, тіофену і піролу зумовлена делокалізацією вільної р-орбіталі гетероатома з утворенням ароматичного секстету електронів. При цьому гетероатом частково лишається нуклеофільних властивостей – здатності до окиснення, утворенню комплексів, алкілуванню.
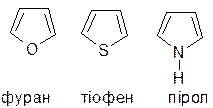
|
|
|
|
|
Дата добавления: 2014-01-05; Просмотров: 3057; Нарушение авторских прав?; Мы поможем в написании вашей работы!