КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Фенантрен
|
|
|
|
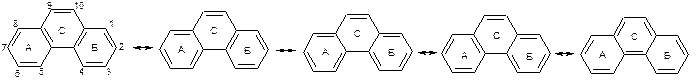
ХІМІЧНІ ВЛАСТИВОСТІ
І. Реакції приєднання
а) Гідрування: 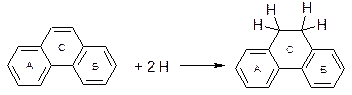
б) Галогенування: 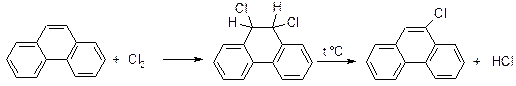
ІІ. Окиснення: 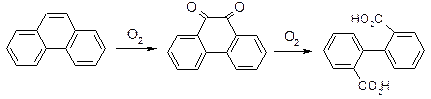
ІІІ. Реакції заміщення: нітрування та сульфування йдуть в положення 9 по тим же причинам, що й заміщення нафталіну в α-положення.
ЕЛЕМЕНТОРГАНІЧНІ СПОЛУКИ
Елементоорганічними сполуками називають такі органічні сполуки, в молекулах яких крім С, Н, О, N, S і галогенів містяться інші елементи Періодичної системи, атоми яких безпосередньо сполучені з атомом карбону.
Елементоорганічні сполуки класифікують за природою елемента. У зв’язку з цим розрізняють Na, Mg, Al, Si, P – та інші елементоорганічні сполуки.
Назва елементоорганічних сполук складається з назви елемента і назви сполученого з ним вуглеводневого радикала, наприклад: - метил натрій, триетилалюміній і т.д.
Всі елементорганічні сполуки підрозділяються на два класи – металоорганічні сполуки (МОС), тобто сполуки, які мають безпосередній зв’язок карбону з металом, та елементоорганічні сполуки (ЕОС), тобто сполуки, які мають безпосередній зв’язок карбону з неметалами, іншими ніж О, N, S та галогени.
В залежності від електронегативності елементів зв’язок карбон-елемент може мати різний характери, причому електронна густина в залежності від відносної електронегативності атомів карбону та елементу може бути зміщена як до, так і від атому елементу. Так, в елементоорганічних сполуках (ЕОС) зв’язок карбон-елемент має ковалентний характер, тоді як в МОС зв’язок може бути трьох типів – дійсно іонний зв’язок (якщо його утворюють найбільш електронегативні метали, такі як Na, K), високо полярний зв’язок із значною часткою іонності (якщо його утворюють інші метали І, ІІ і, частково, ІІІ групи, які мають порівняно низьку електронегативність) та дійсно ковалентний полярний зв’язок (якщо його утворюють всі інші метали). Оскільки карбон більш електронегативний ні, ніж метали, то електронна густина в молекулах МОС завжди зміщена до атому карбону.
Слід пам’ятати, що реакційна здатність зв’язку карбон-метил зростає із зменшенням електронегативності металу, тобто із збільшенням полярності та іонности зв’язку.
Фізичні властивості МОС також залежать від типу зв’язку. Сполуки з іонним зв’язком (наприклад, С-Na) – це тверді солеподібні нелетючі сполуки. Сполуки з ковалентним зв’язком (С-Ме або С-Е: (С2Н5)4Pb або (СН3)4Si) – являють собою рідини або гази, які мають всі типові властивості органічних сполук.
Загальні методи одержання
1. Реакції елементів з галоген алканами (див. галогеналкани).
2. Реакції МОС з галогені дами більш електронегативних елементів:
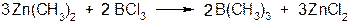
3. Реакції МОС з більш електронегативними металами:
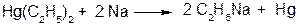
4. Реакції МОС з більш сильними С-Н кислотами:
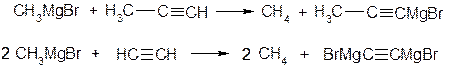
Рушійною силою всіх реакцій є утворення більш типової іонної сполуки (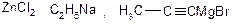 ) та більш типової ковалентнозв’язаної органічної сполуки
) та більш типової ковалентнозв’язаної органічної сполуки 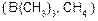 .
.
Як правил, всі МОС добувають у відсутності кисню та води.
Хімічні властивості МОС (загальні реакції)
Магнійорганічні сполуки
І. Розщеплення простих зв’язків
1. Реакції із сполуками, які мають активній (більш кислий) водень
(реакція Чугуєва-Черевітінова)
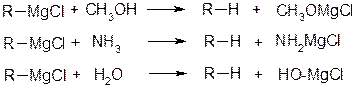
2. Реакції з електронегативними елементами (О, S, Se, Te, VII-група)
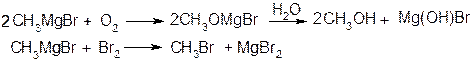
3. Реакції з галогеналкілами
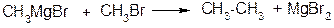
ІІ. Приєднання по подвійним зв’язкам
1. Приєднання по карбонільній групі – утворення спиртів
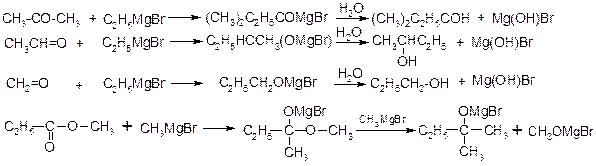
2. Взаємодія з двоокисом вуглецю – утворення кислот
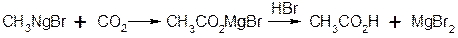
3. Взаємодія з нітрилами – утворення кетонів
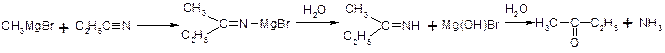
Кремнійорганічні сполуки
Енергія зв’язку Si-Si менша від енергії зв’язку С-С на 125 кДж/моль, тоді як енергія зв’язку С-О менша від енергії зв’язку SiO на 92,1 кДж/моль. Тому для кремнійорганічних сполук характерні не зв’язки Si-Si, а зв’язки Si-О-Si:
1. Алкілхлорсилани, алкілсилани.
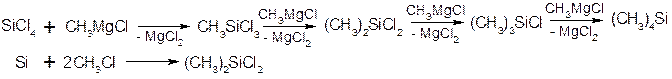
2. Силаноли, силоксани, силікони.
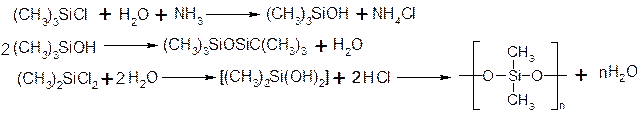
Фосфорорганічні сполуки
1. Фосфіни:
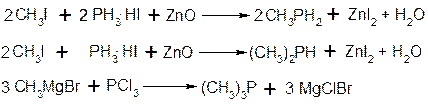
2. Окиснення:
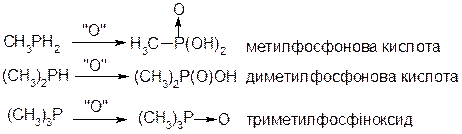
3. Перегрупування Арбузова:
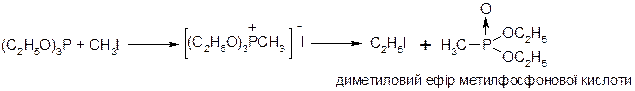
Свинецьорганічні сполуки
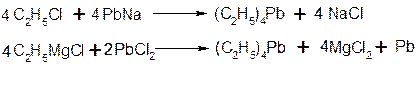
ГАЛОГЕНПОХІДНІ ВУГЛЕВОДНІВ
Галогенпохідними називають сполуки, які містять зв’язки карбон-галоген. Поділяють на
| Аліфатичні | Ароматичні | ||
 Насичені
Насичені
| Ненасичені | Галоген в ядрі | Галоген в боковому ланцюзі |
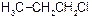 алкілгалогеніди
алкілгалогеніди
| 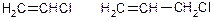 Вініл- та алілгалогеніди
Вініл- та алілгалогеніди
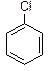
| 
| 
|
В свою чергу любий із типів галогенпохідних може мати 1, 2 і більше атомів галогену, тобто підрозділяють на моно-, ді-, три- і взагалі полігалогенпохідні. Галгенами можуть бути атоми F, Cl, Br, I.
Методи добування моногалогенпохідних
І. Галогенпохідні алканів.
1. Галогенування алканів(див. тему “Алкани”).
2. Приєднання галогеноводнів до алкенів (див. тему “Алкени”).
3. Заміщення гідроксильної групи (-ОН) в молекулах спиртів на галоген:
a. Взаємодія спиртів з галогеноводнями:
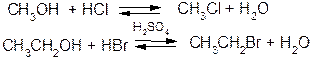
b. Взаємодія спиртів з галоїдними сполуками фосфору:


c. Взаємодія спиртів з хлористим тіонілом (SOCl2):

4. Обмін галогенів

ІІ. Галогенпохідні алкенів.
А. З атомами галогенів у подвійного зв’язку – вінілгалогеніди.
1. Приєднання галогенів до ацетиленових сполук (Дивись - Ацетилени)
2. Відщеплення галогеноводнів від гемінальних та віцинальних дигалогенідів
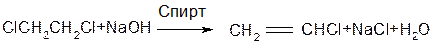
Б. Алілгалогеніди.
1. Алільне галогенування алкенів (Дивись - Алкени)
2. Приєднання галогеноводнів до супряжених дієнів (Дивись - Дієни)
ІІІ. Галогенування бокового ланцюга алкілароматичних вуглеводнів.
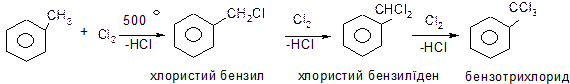
Механізм:
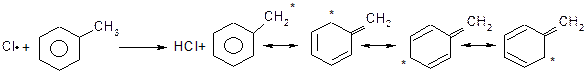
Оскільки бензильний радикал стійкіший за інші можливі за рахунок ефективної делокалізації неспареного електрону з-за супряження з ароматичним кільцем, галогенування любих алкілбензолів завжди проходить лише по α-атому вуглецю бокового ланцюга:
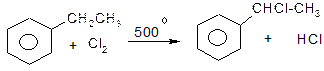
ІV. Галогенування ароматичних сполук в ядро.
Проходить лише каталітично:

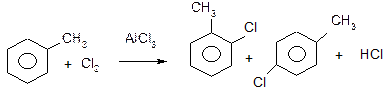
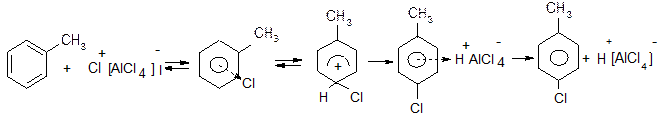
Хімічні властивості
Галогенопохідні – це один з найбільш реакційно здатних класів органічних сполук. Для них найбільш характерні реакції нуклеофільного заміщення (SN) та відщеплення (Е).
І. Реакції нуклеофільного заміщення.
Нуклеофіли – це багаті електронами реагенти, які мають основні властивості. Всі нуклеофіли мають щонайменше одну неподілену пару електронів і характеризуються поняттям нуклеофільність, як основи – основність. Нуклеофільність – це здатність нуклеофільної пари нуклеофілу взаємодіяти з атомом карбону, тоді як основність – це здатність електронної пари основи взаємодіяти з протоном:
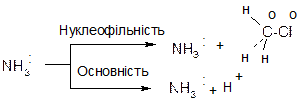
Як правило, нуклеофільність і основність змінюються паралельно, тобто із зростанням основності збільшується нуклеофільність і навпаки. Однак, ці поняття не тотожні і не пропорційні.
При нуклеофільному заміщенні нуклеофіл атакує молекулу, віддаючи для утворення нового зв’язку свою пару електронів. Електрони старого зв’язку, що розривається, С-Наl відходять разом з атомом галогену у вигляді аніона. Оскільки іон галогену – дуже слабка основа (і слабкий нуклеофіл) він легко заміщується будь-якими більш сильними нуклеофілами. Іон галогену є хорошою заміщаючою групою.
У загальному вигляді нуклеофільне заміщення можно забразити такою схемою:
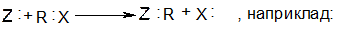
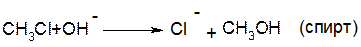
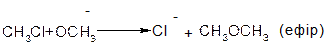
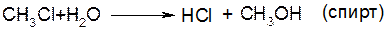
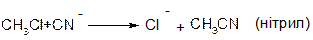
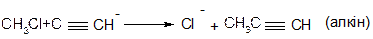
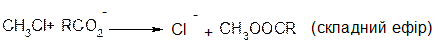
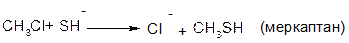
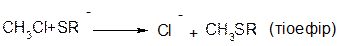

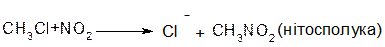
В аліфатичних галогенпохідних, а також в аліл- та бензилгалогенпохідних реакції відбуваються за механізмом SN1 та SN2.
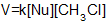 |
Механізм SN2 (заміщення нуклеофільне другого порядку), в якому на лімітуючій стадії реакцій (найменш швидкій стадії, яка визначає швидкість реакції беруть участь два реагенти, тобто)
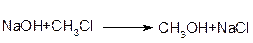
Механізм:
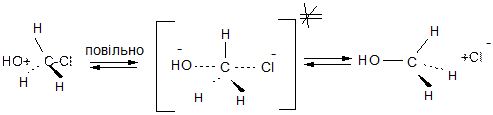
Для SN2 реакцій характерні такі ознаки:
1. Швидкість реакції залежить від концентрації обох реагентів;
2. Реакції супроводжуються, як правило, інверсією конфігурації атому карбону, тобто конфігурація атому карбону в процесі реакції змінюється на протилежну – відбувається Вальденівське обертання конфігурації;
3. Оскільки в реакціях SN2 атака нуклеофілу відбувається зі сторони, протилежної зв’язку С-Hal, швидкість реакції в основному залежить від просторового (стеричного) фрагменту – із збільшенням просторових утруднень атаці нуклеофілу швидкість реакції зменшується:
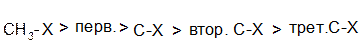
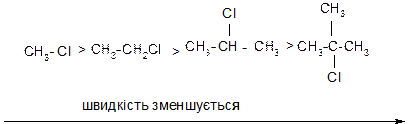
4. При SN2 реакціях, як правило, відсутні перегрупування.
ІІ. Механізм SN1 (заміщення нуклеофільне мономолекулярне),
в якому на лімітуючій стадії приймає участь лише один реагент – галоїдний алкіл, тобто
V=k[R-Hal]:
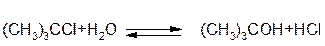

Перша стадія реакції – дисоціація – є лімітуючою стадією. Розрив зв’язку С-Hal відбувається за рахунок енергії сольватації галоген алкану розчинником. Друга стадія відбувається миттєво.
Особливості реакцій SN1:
1. Швидкість реакції залежить лише від концентрації галогену.
2. Реакції, як правило, супроводжуються повною рацемізацією оптично активних сполук, тобто із сполук з визначеною конфігурацією з рівною вірогідністю утворюються сполуки з протилежними конфігураціями. Це відбувається тому, що плоский проміжний карбокатіон з однаковою вірогідністю може атакуватися нуклеофілом з обох сторін.
3. Оскільки в реакціях SN1 на швидкість лімітуючої стадії утворюється карбокатіон, швидкість реакції перед усім залежить від відносної стійкості карбокатіону, тобто від електронних факторів: чим стійкіший утворений карбокатіон, тим більша швидкість реакції. Тому швидкість реакції за механізмом SN1 зменшується в такому ряду:
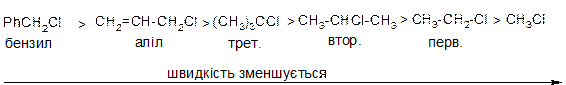
4. Оскільки реакції SN1 відбуваються через проміжне утворення
карбокатіону, вони як правило, супроводжуються перегрупуванням:
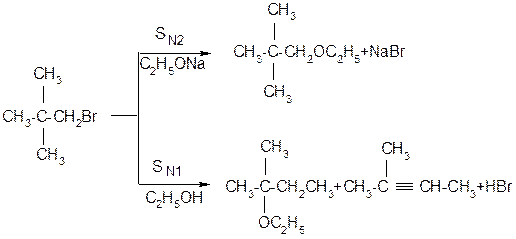
ІІІ. Порівняння реакцій SN1 та SN2.
Нуклеофільне заміщення SN1 супроводжується рацемізацією та перегрупуванням, а реакційна здатність зменшується від третинних до первинних галагенопохідних. Для SN2 реакцій характерне повне обертання конфігурації, відсутність перегрупування, а реакційна здатність зменшується від первинних до третинних галагенопохідних. Оскільки порядок зміщення реакційної здатності в реакціях SN1 та SN2 різна, кожна з них зустрічається в чистому вигляді дуже рідко, в дійсності спостерігається комбінація обох реакцій з більшою або меншою часткою кожної з них. Тому для галогеналканів типовим є присутність мінімуму в реакційній здатності:
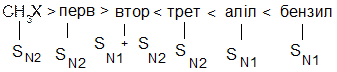
Відзначимо загальні фактори, які впливають на механізм, по якому відбуваються реакції нуклеофільного заміщення.
1. Будова галогеналканів: із збільшенням числа замісників у атома карбону, який несе атом галогену, полегшується реакція за механізмом SN1 та утруднюється реакція за механізмом SN2.
2. Природа залишаючої групи (нуклеофугу). В ряду FclBrI швидкість реакції за обома механізмами зростає, що обумовлено більш легкою іонізацією зв’язку С-Hal з-за зростання його довжини (полегшується реакція за механізмом SN1) та зменшується енергія розпушуючої орбіталі σ*С-Hal (полегшується реакція за механізмом SN2).
3. Концентрація нуклеофільного реагенту: із збільшенням концентрації нуклеофілу полегшується реакція за механізмом SN2 та утруднюється реакція за механізмом SN1.
4. Сила нуклеофільного реагенту: збільшення нуклеофільності реагену сприяє механізму SN2 та протидіє механізму SN1:
5. Природа розчинника. Протонні розчинники сприяють реакціям за механізмом SN1, так як краще соль ватують залишаючи групу (нуклеофуг) і уповільнюють реакції за механізмом SN2, так як зменшують нуклеофільнітьь нуклеофілу за рахунок його сольватації. Апротонні розчинники сприяють реакції за механізмом SN2, так як менше соль ватують нуклеофіли. Але в цілому, полярні розчинники прискорюють реакції SN1, так як перехідний стан цієї реакції більш полярний, ніж вихідний і тому за рахунок сольватації більш стабілізується перехідний стан менш молярний ніж вихідний, вихідний стан більше соль ватується полярними розчинниками і вони відповідно уповільнюють реакції SN2.
ІV. Причини підвищеної реакційної здатності аліл- та бензилгалогенідів в реакціях SN.
Хлористий аліл та хлористий бензил дуже легко вступають в реакції нуклеофільного заміщення за механізмом SN1 із-за високої стійкості проміжних карб катіонів внаслідок ефективного де локалізації позитивного заряду:
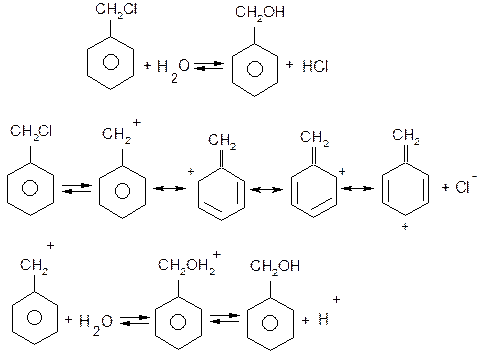
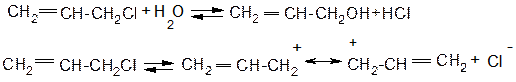
Для алілгалогенідів характерине алільне перегрупування. Так як спряжений алільний катіон здатний приєднувати нуклеофіл за обома атомами карбону, які несуть позитивний заряд:
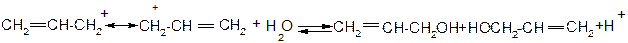
V. Причини інертності атому галогену в вінілгалогенідах та неактивованих арилгалогенідах.
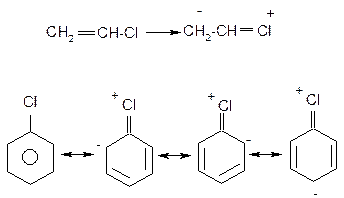
На відміну від розглянутих вище аліл- та бензилгалогенідах в присутності атому галогену безпосередньо при атомі карбону подвійного зв’язку в вініл- та неактивованих арилгалогенідах спряження неподіленої пари електронів атому галогену з сусіднім π- зв’язком вінілгалогенідів та π-системою неактивованих арилгалогенідів, яке приводить до відповідного скорочення зв’язку С-Hal, зв’язок набуває частково подвійний характер та стає більш міцним:
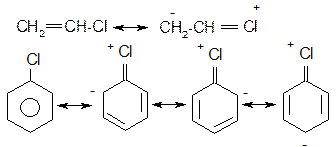
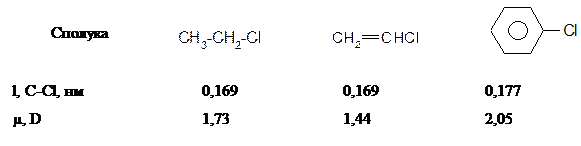 |
В результаті вінілгалогеніди не вступають в реакції нуклеофільного заміщення, а лише в реакції відщеплення; неактивовані арилгалогеніди вступають в реакції псевдо заміщення за механізмом відщеплення-приєднання
VI. Механізм відщеплення – приєднання в неактивованих арилгалогенідах.
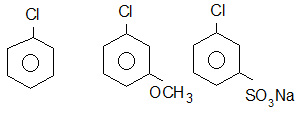 |
До неактивованих арилгалогенідів відносять арилгалогеніди, які не мають з амісників в бензольному ядрі, які мають електронодонорні замісники (замісники І роду), а також частково арилгалогеніди, які мають електроноакцепторні замісники, які знаходяться в м- положеннях відносно атому галогену.
Реакції неактивованих арилгалогенідів відбуваються в дуже жорстких умовах:
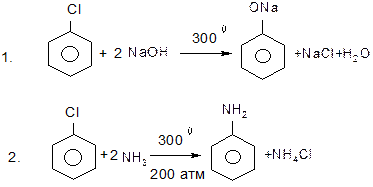
Першою стадією реакції є атака такої основи, як водний розчин лугу або амід натрію, найбільш “кислого” атому гідрогену в бензольному кільці, який знаходиться в о-положенні відносно атому галогену.
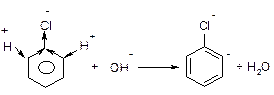
Утворений аніон внаслідок взаємодії вільної пари аніонного центру на атомі карбону з розпушуючою орбіта ллю сусіднього зв’язку С-Hal () викидає аніон хлору з утворенням надзвичайно реакційно здатного дегідробензолу.
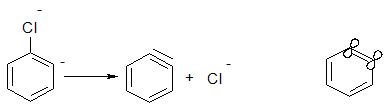
Дегідробензол має ароматичний характер, але, водночас, і потрійний зв’язок. При цьому, одна пара електронів потрійного зв’язку входить до π-електронного секстету бензольного кільця, а інша пара електронів утворює “π- зв’язок”, який розташований в площині бензольного кільця. Цей зв’язок надзвичайно реакційноздатний внаслідок слабкого перекривання двох sp2-гібридизованих електронних орбіта лей і легко вступає в реакції нуклеофільного приєднання (третю стадію реакції):

Місце входження нуклеофілу в похідних дегідробензолу, а також, і в заміщенних неактивованих арилгалогенідах визначається електронними факторами, які діють в похідних дегідробензолу, а не місцем знаходження атому галогену:

Слід пам’ятати, що вплив атому галогену не реакційну здатність неактивованих арилгалогенідів обернений його впливу на реакційну здатність алкілгалгенідів. Так, якщо реакційна здатність алкілгалогенідів зменшується в ряд, то в арилгалогенідах вона зменшується в протилежному напрямку:, що пов’язано з електроноакцепторними властивостями атомів галогену. Наприклад, атом F більш електроноакцепторний від атому Cl, тому в першому випадку ми маємо більш “кислий”, а відповідно, і більш рухливий атом гідрогену в о-положенні по відношенню до атому F.
VII. Активовані арилгалгеніди. Механізм SNAr.
Активовані арилгалогеніди – це арилгалогеніди, які мають в о- або п- положеннях по відношенню до атому галогену електроноакцепторниі замісники, які полегшують атаку нуклеофілу по атому карбону зв’язку С-Hal внаслідок зростання на цьому атомі частково позитивного заряду в результаті прояву ефекту супряження.
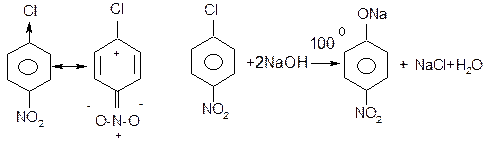
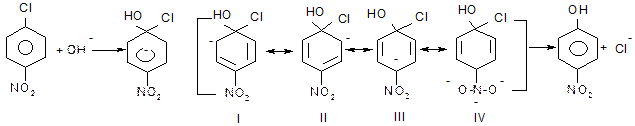
Проміжний негативно заряджений σ-комплекс стабілізується за рахунок ефективного спряження. Найбільший внесок в стабілізацію σ-комплексу вносить межова структура IV, в якій негативний заряд виноситься за межі бензольного кільця і знаходиться на найбільш електронегативному атомі оксигену. Саме утворення енергетично вигідного σ-комплексу під впливом сильного електроноакцепторного замісника є вирішальним фактором здійснення реакції за механізмом SNAr.
Для реакцій SNArшвидкість реакції зростає із зростанням електронегативності атому галогену, тобто зростає позитивний заряд на атомі карбону, який несе атом галогену.
Загалом, вплив атому галогену на швидкість реакцій галагенопохідних можна підсумувати в такій таблиці:
| SN1 | SN2 | E-A | SN-Ar |
| Із зростанням електронегативності атомів галогену швидкість відповідних реакцій | |||
| уповільнюється внаслідок | прискорюється внаслідок | ||
| утворення більш міцного зв’язку С-Hal | збільшення енергії розпушуючої орбіталі σ*C-Hal | появи більш “кислого” атому водню в о-положенні | появи більш позитивного заяду н тоі С, якй несе том Hal |
VIII. Реакції відщеплення галогеноводнів.
Існують два механізми відщеплення галгеноводнів від насичених галогеналканів:
1. Е1 – відщеплення мономолекулярне, швидкість реакції залежить лише від концентраціїгалогеналкану:
V=k[R-Hal]
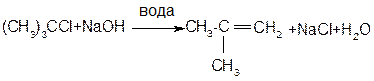
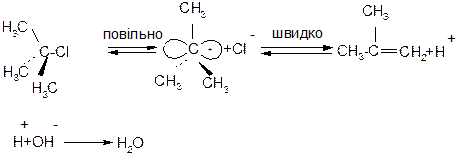
Лімітуючи стадія реакції – утворення карбокатіону, тому швидкість реакції зростає при переході від первинних до третинних алкілгалогенідів:
Відщеплення протону за механізмом Е1 є наглядним проявом ефекту над спряження; протон відщеплюється внаслідок перекривання вакантної орбіталі карбокатіоного центру з електронами сусіднього зв’язку С-Н.
2. Е2 – відщеплення бімолекулярне, швидкість реакції залежить від концентрації обох реагентів – галгеналкану та основи:
V=k[R-Hal][OH-]
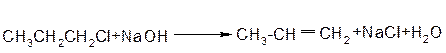
Механізм:
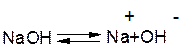
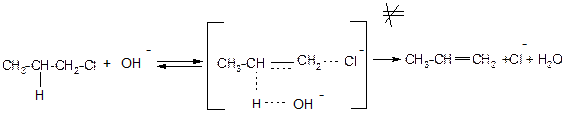
Реакція за механізмом Е2 також прискорюється із збільшенням числа замісників при сусідньому атомі карбону. Це повязоно з утворенням термодинамічно більш вигідного більш розгалуженого Олефіру та меншою міцністю зв’язку С-Н у більш заміщеному атомі карбону.
ІХ. Конкуренція реакцій SN та Е.
Реакції заміщення завжди супроводжуються відщепленням і навпаки. Однак при переході від первинних до третинних галагенопохідних прерд усім полегшуються реакції відщеплення і останні переважають для третинних галагенопохідних. Для третинних галагенопохідних дуже важливо взагалі провести реакцію заміщення. Вона можлива лише тоді, коли використовуються сильні нуклеофіли, які являють собою слабкі нуклеофіли:
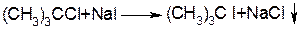
Навпаки, для первинних галагенопохідних переважають реакції заміщення. Вірогідність реакцій відщеплення зростає із збільшенням основності реагентів, в свою чергу реакції заміщення – із збільшенням нуклеофільності реагенту.
Х. Реакції галогеналканів та галогенаренів з металами.
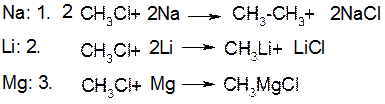
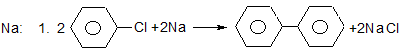
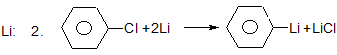
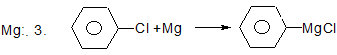
АРОМАТИЧНІ СУЛЬФОКИСЛОТИ
Ароматичними сульфокислотами називають сполуки, в яких атом сірки сульфогрупи –SO3H сполучений з атомом вуглецю ароматичного ядра. У молекулу бензолу можна ввести одну, дві або три сульфогрупи. Прикладом ароматичної моносульфокислоти є бензолсульфокислота:
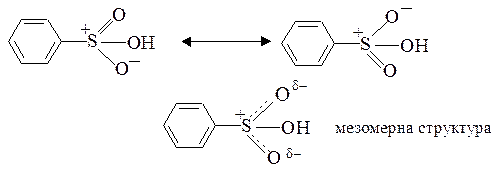
У відповідності до октетної формули сульфогрупа має два ковалентних і два семіполярних зв’язків. Останні утворені за участю тільки електронів атому сірки. Однак, для сульфокислот може використовуватися формула з шістьма ковалентними зв’язками (двома одинарними та двома подвійними), так як атоми сірки мають вільні d-орбіталі, на яких можуть бути розміщені електрони (з розширенням октету).
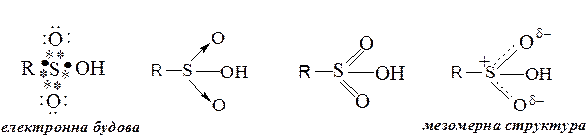
Дійсна будову атому сірки – проміжна між станами, які відповідають цим двом формам (Див. “ мезомерна структура ”).
Ароматичні сульфокислоти добувають переважно реакцією сульфування ароматичних сполук сульфуючими реагентами (концентрована H2SO4, олеум: SO3+H2SO4; SO3; хлорсульфонова кислота HОSO2Cl; хлористий сульфурил SO2Cl2 , та інш.).
Сульфування – це реакція електрофільного заміщення. При сульфуванні сірчаною кислотою сульфуючою частинкою є катіон гідросульфонію, який утворюється з концентрованої кислоти за схемою:
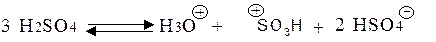
Треба мати на увазі, що реакція сульфування є зворотною:
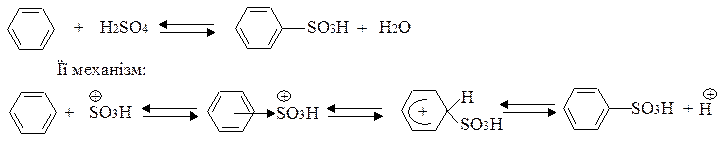 π-комплекс σ-комплекс
π-комплекс σ-комплекс
Подібно до сірчаної кислоти сульфокислоти є сильними кислотами.
Хімічні властивості сульфокислот визначаються наявністю сульфогрупи і їх можна розділити на три типи:
1) реакції сульфогрупи;
2) реакції заміщення сульфогрупи;
3) реакції за участю бензольного ядра.
1. Реакції сульфогрупи
· утворення солей
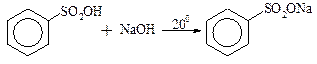
· утворення хлорангідридів
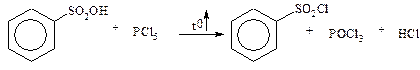
· утворення ефірів та амідів
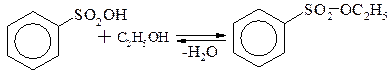
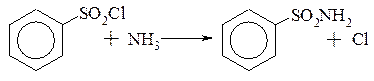
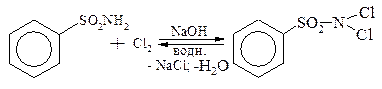
· утворення тіофенолів
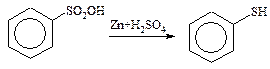
2. Реакції заміщення сульфогрупи на групи –ОН; –CN; –NH2.
· Гідроліз (при дії перегрітої водяної пари в кислому середовищі сульфокислоти дають вихідні ароматичні вуглеводні)
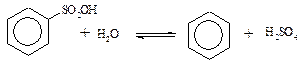
· Реакції лужного плавлення
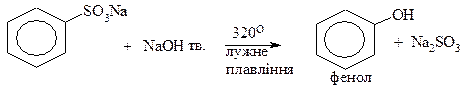
· Сплавлення солей сульфокислот з ціанідами з утворенням нітрилів
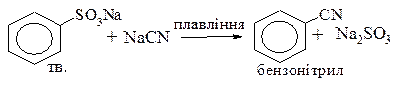
3. Реакції електрофільного заміщення водню: відбуваються важче, ніж у бензолі завдяки тому, що сульфогрупа виявляє негативний індукційний ефект і є орієнтантом другого роду (мета-оріентантом), пасивує бензольне ядро до реакції електрофільного заміщення.

Сульфокислоти мають різноманітне застосування. Вони є основою для добування поверхнево-активних речовин, миючих засобів, барвників, дубильних речовин, йоно-обмінних смол тощо.
ОДНО- І БАГАТОАТОМНІ СПИРТИ
Спирти - це похідні вуглеводнів, у молекулах яких один або декілька атомів водню заміщені гідроксильними групами. У залежності від кількості гідроксильних груп спирти бувають одноатомні та багатоатомні. Молекули спиртів асоційовані внаслідок виникнення між ними водневих зв’язків, чим і пояснюються порівняно високі температури кипіння спиртів:
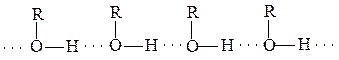
МЕТОДИ ОДЕРЖАННЯ СПИРТІВ
І. Гідроліз галагенопохідних.
а) 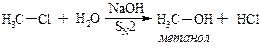
б) 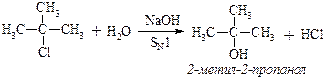
в) Треба мати на увазі, що галоген, який знаходиться біля α-вуглецевого атома бокового ланцюга дуже рухливий і легко заміщується на нуклеофільні реагенти за механізмом SN1:

Спочатку повільно відбувається дисоціація з утворенням енергетично стійкого бензильного катіона, стійкість якого зумовлена спряженням
р -орбіталей бензольного ядра з вакантною орбіталлю вуглецевого атома бокового ланцюга. На другій стадії бензильний катіон швидко реагує з нуклеофілом:
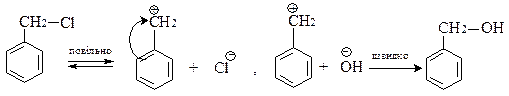
ІІ. Каталітична гідратація алкенів.
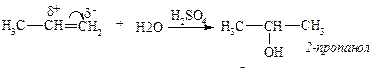
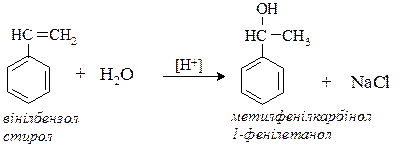
ІІІ. Металоорганічний синтез.
а) для одержання первинних спиртів)

б) для одержання вторинних спиртів
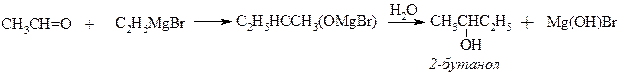
в) для одержання третинних спиртів
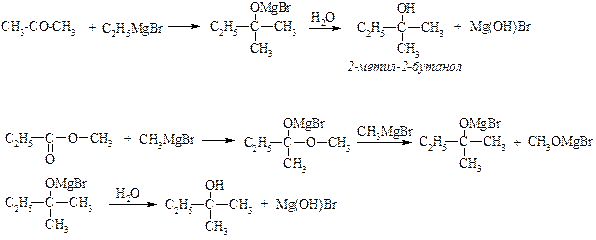
ІV. Гідрування альдегідів та складних ефірів.
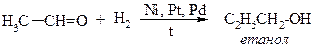
V. Гідрування кетонів.
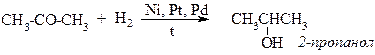
Хімічні властивості спиртів зумовлені присутністю групи –ОН. Спирти – амфотерні сполуки. Виявляють властивості дуже слабких кислот: у реакції з металічним натрієм утворюють солі – алкоголяти:

У реакціях з галогеноводневими кислотами спирти виявляють властивості основ і реагують за механізмами SN2 у первинних спиртів, SN1 у вторинних і третинних. Реакції нуклеофільного заміщення супроводжуються реакціями дегідратації з утворенням алкенів:
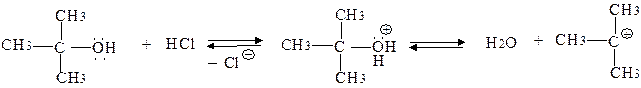
Стабілізація карбокатіону проходить у двох напрямках:
а) приєднання нуклеофіла (1)
б) відщеплення протона (2)
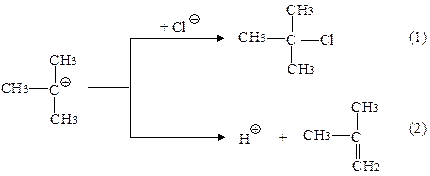
Внутрішньо- та міжмолекулярна дегідратація спиртів
Внутрішньомолекулярна дегідратація спиртів
І) Для вторинних і третинних спиртів за механізмом Е1 (при нагріванні з надлишком сульфатної кислоти за правилом Зайцева)
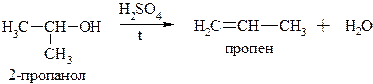
Механізм: Е1
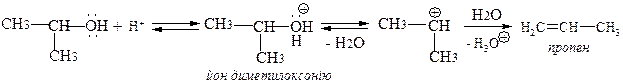
ІІ) для первинних спиртів за механізмом Е2
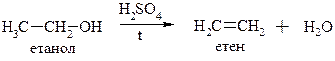
Міжмолекулярна дегідратація спиртів
І) Для вторинних і третинних спиртів за механізмом SN1
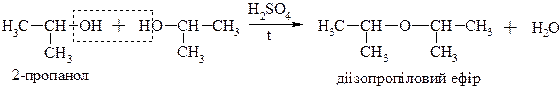
ІІ) для первинних спиртів за механізмом SN2
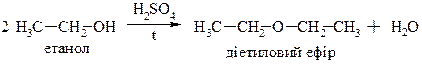
Спирти утворюють також естери при реакції з мінеральними та карбоновими кислотами:

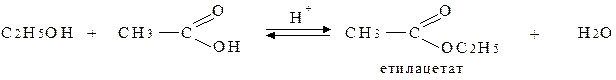
Спирти порівняно легко окиснюються такими окисниками як KMnO4, K2CrO7. Первинні спирти окиснюються до альдегідів, вторинні до кетонів.
Багатоатомні спирти повторюють властивості одноатомних.
Етери, циклічні етери, тіоспирти, тіоетери, сульфокислоти
Етери мають загальну формулу R─O─R’ і утворюються міжмолекулярною дегідратацією спиртів:
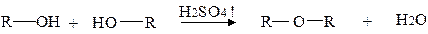
або реакцією алкоголятів з галогеноалкілами:

Етери важко гідролізують. Концентрована сірчана кислота розкладає їх:
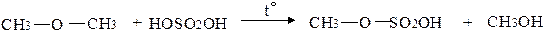
Металевий натрій розщеплює етери при нагріванні:
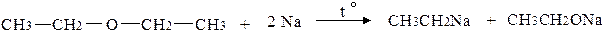
Етери можуть мати циклічну будову. Прикладом може бути оксид етилену 
Для циклічних етерів характерні реакції приєднання сполук, що мають рухливий атом водню:
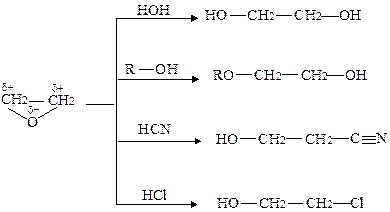
Тіоспирти (тіоли) R─SH і тіоетери (сульфіди) R─S─R’ розглядаються як похідні сірководню.
Тіоспирти виявляють сильніші кислотні властивості ніж звичайні спирти і утворюють солі (тіоляти) в реакції з лугами:
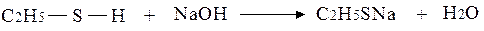
Сульфокислоти – це похідні вуглеводнів, що мають функціональну групу ─SO3H. Їх добувають реакцією сульфоокиснення (1) або сульфохлорування алканів (2) з наступним гідролізом сульфохлориду (3):
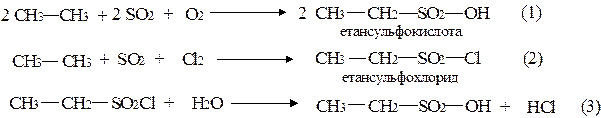 Солі лужних металів вищих алкансульфокислот використовують для одержання пральних засобів.
Солі лужних металів вищих алкансульфокислот використовують для одержання пральних засобів.
ФЕНОЛИ. ХІНОНИ.
МЕТОДИ ОДЕРЖАННЯ ФЕНОЛІВ
І. Гідроліз ароматичних галагенопохідних.
а) неактивованих галагенопохідних
Ці реакції відбуваються за механізмом відщеплення-приєднання (Е-АN). Під дією сильних основних реагентів спочатку від арилгалогеніда відщеплюється галогеноводень. При цьому утворюється надзвичайно реакційноздатний проміжний продукт – дегідробензол, який далі швидко приєднує нуклеофільний реагент:
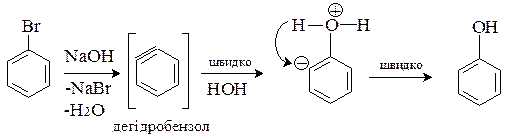
б) активованих галагенопохідних
Активність атома галогену у арилгалогенідах підвищується під впливом електроноакцепторних груп, таких як –NO2, -SO3H та інших, якщо вони знаходяться в о - або п -положеннях відносно атома галогену.
Заміщення відбувається за механізмом SN2Аr через утворення негативно зарядженого σ-комплексу, який стабілізується внаслідок делокалізації негативного заряду електроноакцепторною нітро-групою:
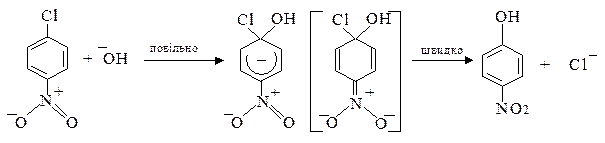 σ -Комплекс Стабільна резонансна
σ -Комплекс Стабільна резонансна
структура
У випадку м -нітрохлорбензолу нітрогрупа не бере участі в делокалізації негативного заряду σ-комплексу і тому галоген у цих сполуках менш активний.
ІІ.Лужне плавення натрієвих солей ароматичних сульфокислот із лугами.
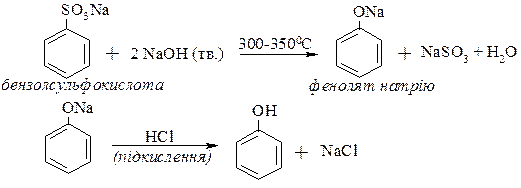
ІІІ. Розклад солей діазонію.
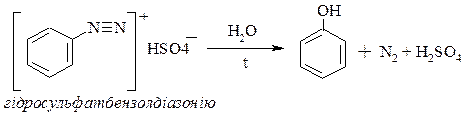
ІV.Кумольний метод (алкілування бензолу за Фріделем-Крафтсом).
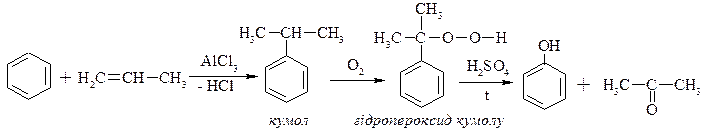
Фенолами називають органічні сполуки, які містять гідроксильну групу, безпосередньо сполучену з вуглецевим атомом бензольного ядра. Залежно від кількості гідроксильних груп в їх структурі феноли поділяють на одноатомні, двоатомні і т. інш.
Хімічні властивості фенолів визначаються гідроксильною групою і сполученим з нею бензольним ядром. Як відомо, гідроксил виявляє –І – ефект і разом з тим вільні пари р-електронів атома кисню взаємодіють з π-електронами бензольного ядра (явище р, π - спряження), тому в результаті такої взаємодії р-електронна густина кисню зміщується в напрямку бензольного ядра (ефект спряження сильніший ніж індукційний).
Внаслідок спряження електронна густина на атомі кисню у молекулі фенолу зменшується, він сильніше притягує до себе електронну густину від атома водню, ніж в аліфатичних спиртах. Це приводить до збільшення полярності О-Н зв’язку і посилення кислотних властивостей фенолів у порівнянні зі спиртами. Це наочно демонструє порівняння констант іонізації (Ка):
pечовина CH3COOH H2CO3 C6H5OH H2O C2H5OH
Ка 1,76·10-5 4,9·10-7 1,3·10-10 1,8·10-16 1·10-18
Таким чином, феноли виявляють властивості слабких кислот і утворюють солі з лугами:
 ,
,
але навіть вугільна кислота витісняє фенол з розчину феноляту:

Електроноакцепторні замісники особливо в орто- і пара-положеннях, значно збільшують кислотність фенолів. Наприклад, тринітрофенол (пікринова кислота) має константу іонізації 1,6·10-1.
За участю гідроксильної групи феноли алкілуються та ацилуються з утворенням простих і складних ефірів. Внаслідок р, π-спряження нуклеофільне заміщення -ОН – групи практично не відбувається, реакція етерифікації також не проходить.
Найважливішими реакціями за участю ароматичного ядра є реакції електрофільного заміщення: галогенування, нітрування, сульфування, алкілування, азосполучення. Вони відбуваються надзвичайно легко в орто- і пара-положеннях ароматичного ядра відносно групи –ОН, внаслідок значного збільшення електронної густини в цих положеннях за рахунок р, π-спряження. Так фенол нітрується 20% розчином азотної кислоти при 10-150С з утворенням суміші орто- і пара-нітрофенолів.
ОСОБЛИВОСТІ РЕАКЦІЙ ЕЛЕКТРОФІЛЬНОГО ЗАМІЩЕННЯ ЗА АРОМАТИЧНИМ ЯДРОМ
Гідроксильна група у фенолів є одним з самих сильних донорів електронів. Вона настільки підвищує електронну густину у бензольному ядрі, що всі реакції електрофільного заміщення проходять у більш м’яких умовах у відсутності каталізатору і з більшою швидкістю ніж у бензолі, крім того у водному розчині фенол дисоціює з виділенням протону і утворенням фенолят аніону:
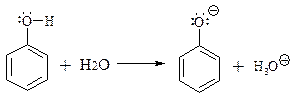
|
|
|
|
|
Дата добавления: 2014-01-05; Просмотров: 2933; Нарушение авторских прав?; Мы поможем в написании вашей работы!