КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Характеристика вагонов 81-717(714) 34 страница
|
|
|
|
Рассмотрим состояние а-орбиталей центрального иона. В свободном ионе электроны, находящиеся на каждой из пяти а-орбиталей, обладают одинаковой энергией (рис. 160, а). Представим себе, что лиганды создают равномерное сферическое электростатическое поле, в центре которого находится центральный ион. В этом гипотетическом случае энергия d-орбиталей за счет отталкивающего действия лигандов возрастает на одинаковую величину, т. е. все d-орбитали останутся энергетически равноценными (рис. 160,6). В действительности, однако, лиганды неодинаково действуют на различные d-орбитали: если орбиталь расположена близко к лиганду, энергия занимающего ее электрона возрастает более значительно, чем в том случае, когда орбиталь удалена от лигаида. Например, при октаэдрическом расположении лигандов вокруг центрального иона наибольшее отталкивание испытывают электроны, находящиеся на орбиталях dz> и dx>-y', направленных к ли-гандам (рис. 161, а и б); поэтому их энергия будет более высокой, чем в гипотетическом сферическом поле. Напротив, dxy, dxz и dyz-орбитали направлены между лигандами (рис. 161, в), так что энергия находящихся здесь электронов будет ниже, чем в сферическом поле. Таким образом, в октаэдрическом поле лигандов происходит расщепление d-уровня центрального иона на два энергетических уровня (рис. 160, в): более высокий уровень, соответствующий орбиталям dz- и dx*-vi (их принято обозначать dy или ъё), и более низкий уровень, отвечающий орбиталям dxy, йхг и dyz (эти орбитали обозначают de или t2g).
Разница в энергиях уровней dy и de, называемая энергией расщепления, обозначается буквой А; ее можно эксперимен-
В dzt dxt.yt
/ г *dv
ту^ iyz diidxfyt
\d*y dxz
1JFa
I dxidyzfydJLy
Рис. 160. Схема энергетических уровней d-орбиталей центрального иона:
а — свободный ион; б — ион в гипотетическом сферическом поле; в — ион в октаэдрическом поле лигандов.
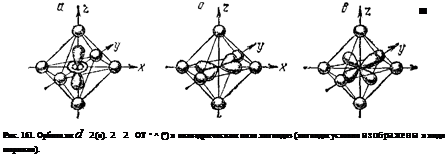
тально определить по спектрам поглощения комплексных соединений. Значение А зависит как от природы центрального атома, так и от природы лигандов: лиганды, создающие сильное поле, вызывают большее расщепление энергетических уровней, т. е. более высокое значение А.
По величине энергии расщепления лиганды располагаются в следующем порядке (так называемый спектрохимический
ряд*):
СО, CN" > этилендиамин (En) > NH3 > SCN" > Н20>ОН" > F"> СГ >Br"> I"
сильное поле
среднее поле
слабое поле
В начале этого ряда находятся лиганды, создающие наиболее сильное поле, в конце — создающие слабое поле.
Электроны центрального иона распределяются по d-орбиталям так, чтобы образовалась система с минимальной энергией. Это может быть достигнуто двумя способами: размещением электронов на de-орбиталях, отвечающих более низкой энергии, или равномерным распределением их по всем d-орбнталям, в соответствии с правилом Хунда (см. § 32). Если общее число электронов, находящихся иа d-орбнталях центрального иона, не превышает трех, то они размещаются на орбнталях более низкого энергетического уровня ds по правилу Хунда. Так, у нона Cr3f, имеющего электронную конфигурацию внешнего слоя 3d3, каждый из трех d-электро-нов занимает одну из трех с?е-орбиталей.
| * Взаимное расположение лигандоз с близкими энергиями расщепления может несколько изменяться прн переходе к другому центральному атому или даже при изменении степени окисленности центрального атома. |
Иное положение складывается, когда на d-орбиталях центрального иона находится большее число электронов. Размещение их в соответствии с правилом Хунда требует затраты энергии для перевода некоторых электронов на <а(у-орбитали. С другой стороны, при размещении максимального числа электронов на с/е-орбиталяк нарушается правило Хунда и, следовательно, необходима затрата
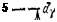
Рис. IC2. Распределение электронов иона Со3+ по d-орби-та ля м:
 |
а
Ц -f -f 4 -К
д
й — в гипотетическом сферическом поле; б — в слабом октаэдрическом поле лигандоз (комплекс ICoFel3"); в—в сильном октаэдрическом по-
ле лигандов (комплекс ICo(CN)6]3")-
энергии для перевода некоторых электронов на орбитали, на кото-' рых уже имеется по одному электрону. Поэтому в случае слабого поля, т. е. небольшой величины энергии расщепления, энергетически более выгодным оказывается равномерное распределение d-элек-тронов по всем d-орбиталям (в соответствии с правилом Хунда); при этом центральный ион сохраняет высокое значение спина, так что образуется высокоспиновый парамагнитный комплекс. В случае же сильного поля (высокое значение энергии расщепления) энергетически более выгодным будет размещение максимального числа электронов на йе-орбиталях; при этом создается низкоспиновый диамагнитный комплекс.
С этой точки зрения понятно, почему, например, комплекс ([СоРб]3~ парамагнитен, а комплекс [Co(CN)6]3- диамагнитен. Положение лигандов F- и CN- в спектрохимическом ряду (см. выше) показывает, что ионам CN- соответствует значительно более высокая энергия расщепления А, чем ионам F-. Поэтому в рассматриваемых комплексах электроны центрального иона Со3+ распределяются по d-орбиталям так, как это показано на рис. 162: комплекс [CoFe]3- — высокоспиновый, а комплекс [Co(CN)6]3-— низкоспиновый.
Мы рассмотрели теорию кристаллического поля в приложении к комплексам с октаэдрическим расположением (октаэдрической координацией) лигандов. С аналогичных позиций могут быть рассмотрены и свойства комплексов с иной, например тетраэдриче-ской, координацией.
На основе теории кристаллического поля удается объяснить не только магнитные свойства комплексных соединений, но и их специфическую окраску. Так, в комплексе [Ti(H20)6]3+ ион Ti3+ имеет один rf-электрон (электронная конфигурация d1). В нормальном (невозбужденном) состоянии этот электрон находится на одной из rfg-орбиталей, но при затрате некоторой энергии (Д = = 238 кДж/моль) может возбуждаться и переходить на dy-орбиталь. Длина волны света, поглощаемого при этом переходе и соответствующего указанной энергии, равна 500 нм: это и обусловливает фиолетовую окраску комплекса [Ti(H20)6]3+. При таком рассмотрении становится понятным, почему комплексы, образованные ионами Cu+, Ag+, Zn2+ и Сц2+, как правило, бесцветны; эти ионы имеют электронную конфигурацию dK, так что все d-орбитали заполнены и переход электронов с d& на dy-орбитали невозможен. Ион же Си2+ образует окрашенные комплексы: он обладает электронной конфигурацией d9, так что один из йе-электронов может при возбуждении переходить на с^-орбиталь.
Хотя теория кристаллического поля оказалась плодотворной в трактовке магнитных, оптических и некоторых других свойств комплексных соединений, она не смогла объяснить положения лигандов в спектрохимическом ряду, а также сам факт образования некоторых комплексов, например, так называемых «сэндвичевых» соединений — дибензолхрома Сг(СбНб)г, ферроцена Fe(CsH5)2 и их аналогов. Дело в том, что теория кристаллического поля, учитывая влияние лигандов на центральный ион, не принимает во внимание участия электронов лигандов в образовании химических связей с центральным ионом. Поэтому применение теории кристаллического поля ограничено, главным образом, комплексными соединениями с преимущественно ионным характером связи между центральным атомом и лигандами.
Метод валентных связей в приложении к комплексным соединениям базируется на тех же представлениях, что и в простых соединениях (см. §§ 39—44). При этом принимается во внимание, что химические связи, возникающие при комплексооб-разовании имеют донорно-акцепторное происхождение, т. е. образуются за счет неподеленной электронной пары одного из взаимодействующих атомов и свободной орбитали другого атома. Рассмотрим с этих позиций строение некоторых комплексных соединений.
В молекуле аммиака атом азота находится в состоянии «/^-гибридизации, причем на одной из его гибридных орбиталей находится неподеленная электронная пара. Поэтому при донорно-акцепторном взаимодействии молекулы NH3 с ионом Н+ образуется ион NH|> имеющий тетраэдрическую конфигурацию. Аналогично построен комплексный ион [BF]4_: здесь донором электронной пары служит анион F- а акцептором—атом бора в молекуле BF3, обладающий незанятой орбиталыо внешнего электронного слоя и переходящий при комплексообразовании в состояние 5р3-гибридизации.
Такую же геометрическую конфигурацию (тетраэдр) имеют некоторые комплексы элементов подгруппы цинка, например [Zn(NH3)4]2+, [Gd(NH3)4]2+, [Hgl4]2-. Так, в комплексе [Zn(NH3)4]2+ ион цинка предоставляет для электронных пар лигандов (условно показанных на схеме точками) одну 4s- и три,4р-орбитали
|2* |

[Zn(NH3)J!
причем осуществляется 5р3-гибридизация, соответствующая размещению лигандов в вершинах тетраэдра (тетраэдрическая координация).
Ионы d-элементов с четырьмя занятыми d-орбиталямн (Pt2+, Pd2+, Au3+) при координационном числе 4 предоставляют для электронных пар лигандов одну (п — \)d-, одну ns- н две лр-орбиталк, например, в комплексе [PI (NHS) 4] 2+:
Р
| S н | fl | р fl | fl | tl | н | н | fl | |
При этом осуществляется гибридизация dsp2, отвечающая размещению лигандов в вершинах квадрата (квадратная координация). Поэтому такие комплексы, как [Pt(NH3)4] 2+, [PtCl4]2-, обладают структурой плоского квадрата.
Координационному числу 6 соответствует гибридизация d2sp3 и октаэдрическое расположение лигандов. Такая координация имеет место, например, в комплексах платины (IV):
[Pt(KH3)6]
4 +
fl
tl fl fl
fl fl fl
1акая же октаэдрическая координация осуществляется в комплексах [Co(NH3)6]3+, [Fe(CN)6]4-, [RhCle]3" и др.
Координационному числу 2 отвечает гибридизация sp-типа и линейная координация лигандов, например, в комплексе [Ag (NH3)2]+:
| tl | !l | p II | fl | fii | fl | fl | fl | f 1 |
Рассмотренные примеры показывают, что метод ВС успешно объясняет определенные значения координационных чисел и геометрические формы комплексных частиц. Правильно описываются с позиций этого метода и различия в магнитных свойствах (диа-магнитность или парамагнитность) комплексных соединений. Однако некоторые их свойства (например, спектры поглощения) не находят с позиций метода ВС удовлетворительного объяснения. Кроме того, взаимодействие между центральным атомом и лиган-дами в комплексных соединениях не сводится только к передаче электронов от лиганда. Существуют лиганды, которые способны принимать электроны металла па вакантные орбитали, например на свободные d-орбитали (в молекуле PF3 или в иоие SnClj), или на незаполненные разрыхляющие орбитали (в молекулах С2Н(, СО, N0). Такие лиганды называют л-акцепторамн, а связь их с центральным атомом л-дативной. Строение многих из открытых в последнее время комплексных соединений, например «сэндвичс-вых», нельзя объяснить с точки зрения метода ВС.
Метод молекулярных орбиталей. Молекулярные орбитали в комплексных соединениях образуются по тому же принципу и обладают теми же свойствами, что и молекулярные орбитали в двухатомных молекулах (см. § 45). Отличие заключается в том, что в комплексных соединениях МО являются многоцентровыми, делокализованными, подобно тому, что имеет место, например, в молекуле бензола (см. § 167).
Метод МО стал в настоящее время ведущим, наиболее плодотворным в теории строения комплексных соединений. В частности, он успешно объясняет строение и свойства уже упоминавшихся «сэндвичевых» соединений, например Сг(СбНб)г, Fe(C5H5b, в которых центральный атом находится между циклическими органическими молекулами и связан с ними делокализованными многоцентровыми связями. Приложение метода МО к объяснению строения комплексных соединений рассматривается в специальных руководствах.
207. Диссоциация комплексных соединений в растворах. Мы
уже говорили о том, что внутренняя и внешняя сфера комплексного соединения сильно различаются по устойчивости;, частицы, находящиеся во внешней сфере, связаны с комплексным ионом преимущественно электростатическими силами и легко отщепляются в водном растворе.
Эта диссоциация называется первичной, она протекает почти нацело, по типу диссоциации сильных электролитов. Лиганды, находящиеся во внутренней сфере, связаны с центральным атомом значительно прочнее и отщепляются лишь в небольшой степени. Обратимый распад внутренней сферы комплексного соединения носит название вторичной диссоциации. Например, диссоциацию комплекса [Ag(NH3)2]Cl можно записать так:
[Ag(NH3)2]Cl -—>• [Ag(NH3)2]++ СГ — первичная диссоциация [Ag(NH3)2]+ щ=±: Ag+ + 2NH3 — вторичная диссоциация
Вторичная диссоциация характеризуется наличием равновесия между комплексной частицей, центральным ионом и лигандами. В этом можно убедиться на основании следующих реакций. Если на раствор, содержащий комплексный ион [Ag(NH3)2]+, подействовать раствором какого-нибудь хлорида, то осадка не образуется, хотя из растворов обычных солей серебра при добавлении хлоридов выделяется осадок хлорида серебра. Очевидно, концентрация ионов серебра в аммиачном растворе слишком мала, чтобы при введении в него даже избытка хлорид-ионов можно было бы достигнуть значения произведения растворимости хлорида серебра (riPAgci = 1,8-Ю-10). Однако после прибавления к раствору комплекса иодида калия выпадает осадок иодида серебра. Это доказывает, что ионы серебра все же имеются в растворе. Как ни мала их концентрация, но она оказывается достаточной для образования осадка, так как произведение растворимости иодида серебра Agl составляет только 1-10—16, т. е. значительно меньше, чем у хлорида серебра. Точно так же при действии сероводорода получается осадок сульфида серебра Ag2S, произведение растворимости которого равно 10-51.
Уравнение протекающих реакций можно записать так:
[Ag(NH,),]++ Г—> AgI| + 2NH,
2[Ag(NH3)2]+ + H2S—> Ag2Sj + 2NH3 4-2NH4+
'Диссоциация ионов [Ag(NH3)2]+, согласно приведенному выше уравнению, как и диссоциация всякого слабого электролита, подчиняется закону действия масс и может быть охарактеризована соответствующей константой равновесия, называемой константой нестойкости комплексного иона:
к.. [Ag+][NH,]»,..fi, 1Q-8
Константы нестойкости для различных комплексных ионов весьма различны и могут служить мерой устойчивости комплекса. Константы нестойкости, в выражения которых входят концентрации ионов и молекул, называются «концентрационными». Более строгими и не зависящими от концентраций и ионной силы раствора являются константы нестойкости, содержащие вместо концентраций активности ионов и молекул. В разбавленных растворах эти два различных выражения констант нестойкости совпадают друг с другом,
Из приведенной формулы видно, что чем меньше концентрация продуктов распада, т. е. чем устойчивее комплекс, тем меньше его константа нестойкости. Наиболее устойчивые в растворах комплексные частицы имеют наименьшие константы нестойкости. Так,среди однотипных соединений
IAg(N02)2r [Ag(NH3)2l+ [Ag(S203)2]3- |Ag(CN)2]-
/Хнест 1,3 • Ю-3 6,8 - 10~8 1-10-13 МО"21
устойчивость комплекса возрастает при переходе от [Ag(N02)2]~ к [Ag(CN)2]-. Ион [Ag(CN)2]~ настолько стоек, что даже прибавление иодида калия к раствору комплексной соли не приводит к образованию осадка иодида серебра. Но при действии сероводорода, ввиду ничтожно малого значения произведения растворимости сульфида серебра, все же выпадает осадок сульфида серебра.
207. Диссоциация комплексных соеди:
к в раствор
В последнее время для характеристики устойчивости комплексных соединений предпочитают пользоваться величиной, обратной константе нестойкости, называемой константой устойчи» в о с т и. Для иона [Ag(NH3)2]+ константа устойчивости равна:
К 1 ^ [[Ag(NH3)2n
УСТ /Сиест [Ag+] [NH3]2
До сих пор шла речь об общих константах нестойкости и устойчивости, относящихся к распаду комплекса на конечные продукты. В действительности же в растворах имеет место ступенчатая диссоциация комплекса, аналогично ступенчатой диссоциации слабых электролитов, например многоосновных кислот.
Так, в водном растворе Кг [PtCU] присутствуют в различных соотношениях все комплексы, участвующие в равновесиях:
[PtCl4]2- + H20 ^ [PtCl3H20]- + cr
[PtCl3H20]" + Н20 [PtCl2(H20)2] + СГ
[PtCl2(H20)2] + Н20 £ъ [Pt(H20)3Cir + СГ
[Pt(H20)3Cl]+ + H20 £ъ [Pt(H20)4]2++Cr
Каждое из этих равновесий характеризуется своей ступенчатой константой нестойкости /<4, Кз и т. д. По мере отщепления хлорид-ионов заряд комплекса становится все более положительным, а число ионов С1~ в комплексе уменьшается. В результате последовательный отрыв хлорид-ионов все в большей степени затрудняется. Поэтому между ступенчатыми константами нестойкости иона [PtCl4]2- имеет место соотношение: 7(4 > /<3 > К2 > К\.
Такое изменение в значениях последовательных констант нестойкости носит общий характер *. Значение общей константы нестойкости равно произведению всех ступенчатых констант.
Значения констант нестойкости и устойчивости приводятся в справочниках по химии. С помощью этих величин можно предсказать течение реакций между комплексными соединениями: при сильном различии констант устойчивости реакция пойдет в сторону образования комплекса с большей константой устойчивости или, что равноценно, с меньшей константой нестойкости. Например, для иона [Ag(NH3)2]+ Янест = 6,8-Ю-8, а для иона NH + /(«ест = = 5,4-Ю-1; поэтому под действием кислот аммиакат серебра разрушается с образованием ионов Ag+ и NH*:
| * Имеется небольшое число отклонений, вызываемых диспропорционирова-нием или действием дополнительных факторов, приводящих к предпочтительной стабилизации одной из равновесных форм. |
[Ag(NH3)2]++ 2Н+ =«=fc Ag+ + 2NH4+
Комплекс же [Pt(NH3)4]2+ (/Снест = 5-10~34) не разрушается при комнатной температуре даже в концентрированной соляной кислоте.
Процессы комплексообразования широко используются в аналитической химии. При выборе условий наиболее эффективного разделения ионов исходят из соотношения констант устойчивости образуемых ими комплексных соединений.
Например, катионы Ni2+, Со2+, Zn2+ дают устойчивые растворимые аммиакаты, а Al3+, Fe3+, Сг3+ менее склонны к комплексообра-зованпю с аммиаком и осаждаются при действии аммиака в виде гидроксидов. Это позволяет разделить действием аммиака эти две группы катионов. Подобного рода соображения могут быть использованы для разделения анионов: так, можно осадить смесь хлоридов и иодидов в виде AgCl и Agl и далее обработать ее аммиаком— в раствор перейдет только AgCl, a Agl останется в осадке. Для того чтобы растворить Agl, нужно применить лиганд, связывающий ион Ag+ значительно прочнее, например CN-, так как для комплекса [Ag(CN)2]~ Янест = ЬЮ-21. В растворе KCN иодид серебра растворяется с образованием K[Ag(CN)2]:
AgI + 2KCN —► K[Ag(CN)2]+KI
Константы устойчивости однотипных комплексов зависят от ряда факторов, и прежде всего от природы центрального атома и лигандов. В комплексах с центральными ионами, обладающими слабой поляризующей способностью, например с ионами щелочных и щелочноземельных металлов, устойчивость растет по мере увеличения интенсивности электростатического взаимодействия между центральным ионом и лигандами: чем больше заряды центрального иона и лигандов и чем меньше их радиусы, тем выше устойчивость комплексов. Эти катионы образуют более устойчивые комплексы с лигандами, содержащими элементы малых периодов (кислород, азот) и с ионами F-.
Для другой большой группы комплексообразователей — катионов платиновых металлов, ионов Hg2+, Ag+, Au3+, у которых поляризующая способность выражена сильно и характер связи центрального атома с лигандами приближается к ковалентному, — наиболее устойчивы комплексы с легко поляризующимися лигандами. К последним относятся, например, ионы I" и лиганды, содержащие атомы Р и S.
208. Влияние координации на свойства лигандов и центрального атома. Взаимное влияние лигандов. Координация сопряжена с изменением электронной конфигурации лигандов и в результате приводит к изменению их свойств. Это хорошо видно на примере кислотно-основных свойств комплексных соединений. В то время как свободный аммиак обладает в водном растворе основными свойствами, комплекс [Р1(ЫНз)б]4+ проявляет свойства кислоты и вступает в обратимую реакцию со щелочью:
[Pt(NH3)6]4- + OH- zf=± [Pt(NH3)5N:H2]3+ + H20
Причина изменения свойств аммиака заключается в том, что при его координации происходит смещение электронной плотности к положительно заряженному центральному атому. В результате эффективный отрицательный заряд атома азота в молекуле NH3 резко снижается, что и облегчает отщепление протона.
Аналогично ведут себя в поле катионов некоторых переходных металлов и другие полярные или легко поляризующиеся молекулы, способные проявлять протонодонорные свойства — Н2О, NH2OH, органические амины. Выступая в качестве лигандов, они способны к отщеплению протона в водных растворах и с точки зрения протонной теории кислот и оснований (стр. 237) ведут себя как кислоты. Например, взаимодействие гидратнрованного иона меди с водой следует записать так:
[Сн(Н20)4]2++ Н20 [Cu(H20)3OH]++ Н.,0+
Это уравнение выражает процесс гидролиза иона меди. Таким образом, гидролиз катионов в водных растворах можно рассматривать как кислотную диссоциацию воды в аквакомплексах.
Если в комплексном соединении одновременно содержатся протонодонор-ная молекула лигапда (например, Н20, NH3) и молекула того же лиганда, отщепившая протон и способная, следовательно, к его присоединению (например, ОН", NH2), то такое комплексное соединение будет амфотерным. Так, комплекс [Pt(NH3)5NH2]3+ в реакции со щелочью ведет себя как кислота, а в реакции с кислотой — как основание:
lPt(NH3)5NH2]3+ + ОН' [Pt(NH3)4 (NH2)2]2+ + Н20
кислота основание
[Pt(NH3)5NH2]3++ Н30+ ч=* lPt(NH3)6]4++ Н20
основание кислота
Соединение, содержащее только депротонированные ионы (ОН", NH2 NH^O" и т. п.), является уже только основанием.
Основные положения теории кислотно-основных свойств комплексных соединений были разработаны А. А. Гринбергом*.
| * Александр Абрамович Гринберг (!898—1966), крупный советский химик-неорганик, академик, лауреат Государственной премии. Основные труды А. А. Гринберга посвящены изучению пространственного строения, кислотно-основных и окислительно-восстановительных свойств комплексных соединений. Им сделан ряд ценных обобщений о влиянии координации на свойства центрального атома и лигандов, о реакционной способности комплексных соединений, |
Центральный ион также меняет свои свойства в результате ком-плексообразования, что можно видеть, например, по изменению соответствующего электродного потенциала. Так, стандартный электродный потенциал е° системы Fe3+/Fe2+ в водном растворе равен +0,771 В. Если же взять цианидные комплексы, содержащие железо в степени окисленности -j-2 и +3, то для системы [Fe(CN)6]3-/[Fe(CN)6]4- е° = +0,36 В, из чего следует, что эта система обладает более слабыми окислительными свойствами, чем система Fe2+/Fe3+. В данном, наиболее типичном случае переход
от гидратированных ионов к более устойчивым комплексам сопровождается преимущественной стабилизацией комплексного иона, содержащего центральный атом в высшей степени окисленности, вследствие чего окислительная способность этого иона ослабляется.
Кроме влияния комплексообразования на свойства лигандов и центрального атома, существует и взаимное влияние лигандов в комплексах. Наиболее ярким его проявлением является транс-влияние, открытое И. И. Черняевым *. Суть этого явления заключается в том, что в комплексах, для которых возможны цис- и транс-изомеры, взаимное влияние лигандов наиболее сильно проявляется при их размещении в транс-положении по отношению друг к другу.
| [Pt(NH3)4]Cl2 + 2НС1 K2[PtCl4] + 2NH3 - |
Некоторые лиганды (их называют транс-активными) ослабляют воздействие центрального атома па свойства лиганда, находящегося в гране-положении по отношению к рассматриваемому лигапду, и приближают их к свойствам свободного лиганда. Примером проявления транс-влияния может служить получение изомерных диаммипов платины(П). При нагревании тетраамми-нов платины(П) с концентрированной НС1 обычно получаются транс-изомеры, а при действии аммиака на K2[PtCl4] — цис-изомеры:
- [Pt(NH3)2Cl2] + 2NH4C1 транс-нзошер
[Pt(NH3)2Cl2] + 2КС1
^ас-изомер
Это объясняется большим транс-влиянием иона С1~ по сравнению с молекулой NH3, что и определяет наиболее лабильный лиганд (обведен) в промежуточно образующихся комплексах:
|
|
|
|
|
Дата добавления: 2014-11-16; Просмотров: 492; Нарушение авторских прав?; Мы поможем в написании вашей работы!