КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Ol — V2X 16 страница
|
|
|
|
критическая точка
 Изобразим в плоскости Р, V какую-либо изотерму жидкости и газа, т. е. кривую зависимости Р от V при изотермическом расширении однородного тела (abc и def на рис. 18). Согласно термодинамическому неравенству (dPJdV)T <0, Р есть убывающая функция V. Такой наклон изотерм должен сохраниться и на некотором протяжении за точками их пересечения с.кривой равновесия жидкости и газа (точки b и е); участки be и ed изотерм соответствуют метастабильным. перегретой жидкости и пере- " охлажденному пару, в которых термодинамические неравенства по-прежнему соблюдаются (полностью же равновесному изотермическому изменению состояния между точками b и е отвечает, конечно, горизонтальный отрезок be, на котором происходит расслоение на две фазы). Если учесть, что точки b н е имеют одинаковую ординату Р, то ясно, что обе части изотермы не могут перейти друг в друга непрерывным образом, и между ними должен быть разрыв. Изотермы заканчиваются в точках (с и d), в которых нарушается термодинамическое неравенство, т. е.
Изобразим в плоскости Р, V какую-либо изотерму жидкости и газа, т. е. кривую зависимости Р от V при изотермическом расширении однородного тела (abc и def на рис. 18). Согласно термодинамическому неравенству (dPJdV)T <0, Р есть убывающая функция V. Такой наклон изотерм должен сохраниться и на некотором протяжении за точками их пересечения с.кривой равновесия жидкости и газа (точки b и е); участки be и ed изотерм соответствуют метастабильным. перегретой жидкости и пере- " охлажденному пару, в которых термодинамические неравенства по-прежнему соблюдаются (полностью же равновесному изотермическому изменению состояния между точками b и е отвечает, конечно, горизонтальный отрезок be, на котором происходит расслоение на две фазы). Если учесть, что точки b н е имеют одинаковую ординату Р, то ясно, что обе части изотермы не могут перейти друг в друга непрерывным образом, и между ними должен быть разрыв. Изотермы заканчиваются в точках (с и d), в которых нарушается термодинамическое неравенство, т. е.
{w)t=°- (83>!)
| J) Участок изотермы, соответствующий перегретой жидкости (be на рис. 18), может оказаться расположенным частично под осью абсцисс. Другими словами, перегретая жидкость может обладать отрицательным давлением; такая жидкость действует на ограничивающую ее поверхность с силой, направленной внутрь объема жидкости. Таким образом, давление не есть величина непременно положительная, и в природе могут существовать—хотя только как метастабиль-ные—также и состояния тела с отрицательными значениями давления (об этом уже шла речь в § 12). |
Построив геометрическое место точек окончания изотерм жидкости и газа, мы получим кривую АКБ, на которой нарушаются (для однородного тела) термодинамические неравенства; она ограничивает область, в которой тело ни при каких условиях не может существовать как однородное. Области между этой кривой и кривой равновесия фаз отвечают перегретой жидкости и переохлажденному пару1). Очевидно, что в критической точке обе кривые должны касаться друг друга. Из точек же, лежащих на самой кривой АКБ, реально существующим состояниям однородного тела отвечает лишь критическая точка К—единственная, в которой эта кривая соприкасается с областью устойчивых однородных состояний.
Полезно указать, что условие (83,1) в критической точке может быть получено из следующих простых соображений. Вблизи критической точки удельные объемы жидкости и пара близки друг к другу. Обозначив их посредством V и V + напишем условие равенства давлений обеих фаз в виде
Р (V, T) = P(V + 6V, Т). (83,2)
Разложив правую сторону равенства по степеням bV и разделив на малую, но конечную величину 8V, найдем
(£),+¥(£),+••-<>■ <ад
Отсюда видно, что при стремлении 8V к нулю, т. е. в критической точке, (дР/дУ)т во всяком случае должно обратиться в нуль.
В противоположность обычным точкам фазового равновесия критическая точка является в математическом отношении особой точкой для термодинамических функций вещества (то же самое относится ко всей кривой АКВ, ограничивающей область существования однородных состояний тела). Характер этой особенности и поведение вещества вблизи критической точки будут рассмотрены в § 153.
§ 84. Закон соответственных состояний
Интерполяционное уравнение состояния ван-дер-Ваальса
r~V—Nb У2'
находится в качественном соответствии с описанными выше свойствами перехода между жидкостью и паром.
Определяемые этим уравнением изотермы представлены на рис. 19. Кривые, проходящие над критической точкой К, изображают монотонно убывающие функции Р (V) при Т > Гкр. Изотерма, проходящая через критическую точку, имеет в ней перегиб. При температурах же Г < Ткр каждая изотерма имеет минимум и максимум, между которыми лежит участок с (дР/дУ)т>0; эти участки (показанные на рис. 19 пунктиром) не соответствуют каким бы то ни было реально существующим в природе однородным состояниям вещества.
Как уже было объяснено в предыдущем параграфе, равновесному переходу жидкости в газ соответствует горизонтальный прямой отрезок, пересекающий изотерму. Уровень, на котором должен быть проведен этот отрезок, определяется условием фазового равновесия n1 = \ii, которое напишем в виде
Jd\i=О,
где интеграл берется по пути перехода из состояния одной фазы в состояние другой фазы. Интегрируя вдоль изотермы, имеем
 |
d\i = vdP,
так что
(84,2)
Геометрически это условие означает равенство площадей, заштрихованных на рис. 19 для одной из изотерм (правило Максвелла).
Критические температура, давление и объем могут быть выражены через параметры а и Ь, входящие в уравнение ван-дер-Ваальса. Для этого дифференцируем выражение (84,1) и пишем уравнения
дР \ dV)т''
о (Щ =0
определяющие точку перегиба на изотерме. Вместе с уравнением (84,1) они дают
8 а
КР — 27 Т '
| кр 27 о2 |
1 а
V =ЗМ> Р = — —
' Кр, * КО 07 А2
(84,3)
Введем теперь приведенные температуру, давление и объем согласно определениям
f____ 71 р,______ Р у,________ V
1 т > * р > ' и
' Кр г Кр * Кр
(84,4)
Выраженное через эти величины, уравнение ван-дер-Ваальса принимает вид
(Р'+уъ) (ЗУ'-1) = 8Г. (84,5)
В это уравнение входят только У, Я' и Г' и не входят никакие величины, характеризующие данное вещество. Поэтому урав

ГЛАВА IX
РАСТВОРЫ
§ 85. Системы с различными частицами
До сих пор мы ограничивались рассмотрением тел, состоящих из одинаковых частиц. Теперь мы перейдем к исследованию систем, состоящих из различных частиц. Сюда относятся всякого рода смеси нескольких веществ; если одного из веществ в смеси значительно больше, чем других, то такую смесь называют раствором остальных веществ в этом преобладающем (растворителе).
Принято называть числом независимых компонент системы число веществ, количества которых в состоянии полного равновесия могут быть заданы произвольно. Все термодинамические величины системы в полном равновесии вполне определяются, например, значениями температуры, давления и числами частиц независимых компонент. Число независимых компонент может не совпадать с полным числом различных веществ в системе, если между этими веществами может происходить химическая реакция; если такая система находится в неполном равновесии, то для определения ее термодинамических величин необходимо, вообще говоря, задание количеств всех входящих в нее веществ.
Легко обобщить результаты § 24 на тела, состоящие из различных веществ. Прежде всего все термодинамические величины должны быть однородными функциями первого порядка по отношению ко всем аддитивным переменным—числам различных частиц и объему.
Далее, вместо понятия об одном химическом потенциале тела как производной от какого-либо из его термодинамических потенциалов по числу частиц (§ 24), появляются химические потенциалы р,- каждой из компонент смеси — производные от термодинамического потенциала по числам частиц N,- этих компонент. Соответственно во всех формулах (24,5), (24,7—9) вместо члена paW надо писать теперь сумму ^ju^-dAf,-.
Так, выражение для дифференциала а*Ф запишется в виде
d<t> = — S dT + V dP + 2 Р,- dNh

|
| V-t = |
а химический потенциал
(85,1)
Химические потенциалы выражены при этом как функции от давления, температуры и концентраций, т. е. отношений чисел частиц различных веществ. Последние могут входить в и.,- только в виде отношений, так как, поскольку Ф есть однородная функция первого порядка от Nh химические потенциалы должны быть однородными функциями нулевого порядка по этим переменным.
Из того факта, что Ф есть однородная функция первого порядка относительно Nh следует, согласно теореме Эйлера,
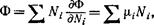
(85,2)

и отсюда опять формулу Q = —PV. Эта последняя теряет свою применимость только для тел, находящихся во внешнем поле, когда давление в разных частях тел различно.
Непосредственно обобщаются также и результаты § 25: условия равновесия системы во внешнем поле требуют постоянства вдоль системы наряду с температурой также и химических потенциалов каждой из компонент:
р,,= const.
(85,3)
Наконец, распределение Гиббса для систем, состоящих из различных частиц, приобретает вид
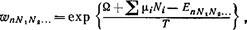
(85,4)
что является естественным обобщением формулы (35,2).
§ 86. Правило фаз
Рассмотрим теперь систему, состоящую из различных веществ и представляющую собой совокупность нескольких (г) соприкасающихся друг с другом фаз (каждая фаза содержит при этом, вообще говоря, все вещества).
Число независимых компонент в системе пусть будет п. Тогда каждая фаза характеризуется давлением, температурой и п хи

г</г + 2.
(86,2)
Другими словами, в системе, состоящей из п независимых компонент может находиться одновременно в равновесии не больше чем п + 2 фазы. Это—так называемое правило фаз Гиббса. Частный случай этого правила мы имели в § 81: в случае одной компоненты число фаз, могущих существовать одновременно, соприкасаясь друг с другом, не может быть больше трех.
Если число г сосуществующих фаз меньше, чем п + 2, то в уравнениях (86,1) п + 2—г переменных могут, очевидно, иметь произвольные значения. Другими словами, можно произвольно менять любые и+ 2—г переменных, не нарушая равновесия; при этом, конечно, остальные переменные меняются совершенно определенным образом. Число переменных, которые могут быть произвольно изменены без нарушения равновесия, называются числом термодинамических степеней свободы системы. Если обозначить его буквой /, то правило фаз можно написать в виде
/ = п + 2-г,
(86,3)
где f не может быть, конечно, меньше нуля. Если число фаз равно своему максимальному возможному значению п + 2, то / = 0, т. е. в уравнениях (86,1) все переменные определены, и ни одной из них нельзя изменить без того, чтобы не нарушилось равновесие и не исчезла какая-нибудь из фаз.
§ 87. Слабые растворы
Мы займемся теперь (§§ 87—91) изучением термодинамических свойств слабых растворов, т. е. таких растворов, в которых число молекул растворенных веществ значительно меньше числа молекул растворителя. Рассмотрим сначала случай раствора с одним растворенным веществом; обобщение для раствора нескольких веществ можно будет произвести непосредственно.
Пусть N—число молекул растворителя в растворе, а п—-число молекул растворяемого вещества. Концентрацией раствора назовем отношение n/N = c; согласно сделанному предположению
Найдем выражение для термодинамического потенциала раствора. Пусть Ф0(Г, Р, N) есть термодинамический потенциал чистого растворителя (в котором ничего не растворено). Согласно формуле Ф = Жр (справедливой для чистых веществ) его можно написать в виде Ф0 = N\ia (Р, Т), где р0 (Р, Г) —химический потенциал чистого растворителя. Обозначим посредством а = а(Р, Т, N) малое изменение, которое испытал бы термодинамический потенциал при введении в растворитель одной молекулы растворяемого вещества. В силу предполагаемой слабости раствора молекулы растворенного вещества в нем находятся на сравнительно больших расстояниях друг от друга, и потому их взаимодействие слабо. Пренебрегая этим взаимодействием, можно утверждать, что изменение термодинамического потенциала при введении в растворитель п молекул растворяемого вещества равно па. Однако в получаемом таким путем выражении Ф0 + па еще не учтена должным образом одинаковость всех молекул растворенного вещества. Это есть выражение, которое получилось бы по формуле (31,5), если бы при вычислении статистического интеграла все частицы растворенного вещества считались отличными друг от друга. Как мы знаем (ср. (31,7)), вычисленный таким образом статистический интеграл должен в действительности еще быть поделен на п \1).
Это приводит к появлению в свободной энергии, а потому и в потенциале Ф дополнительного члена Г In и!. Таким образом,
Ф = Л/р0(Р, Т)+па(Р, Т, N) + T\nn\.
а) Мы пренебрегаем здесь квантовыми эффектами, что для слабого раствора—как и для достаточно разреженного газа—всегда законно.
§ 87]
слабые растворы
Далее, поскольку п—само по себе очень большое число, хотя и малое по сравнению с N, в последнем члене можно заменить In я! = п In (п/е). Тогда
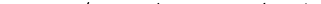 |
Учтем теперь, что Ф должно быть однородной функцией первого порядка по отношению к п и N. Для этого, очевидно, стоящая под знаком логарифма функция ехр (а/Т) должна иметь вид f(P, T)/N. Таким образом,
Ф = ЛГц.0 + лПп [^f(P, Т)].
Вводя новую функцию от Р и Т: ty(P, Т) = Т In f(P, Т), находим окончательно для термодинамического потенциала раствора выражение
Ф = ЛГМР, T)+nTln^ + n$(P, Т).
(87,1)
Сделанное в начале этого параграфа предположение относительно прибавления члена вида па к потенциалу чистого растворителя есть в сущности не что иное, как разложение в ряд по степеням п с оставлением только первых членов. Член следующего порядка по п пропорционален яа, а с учетом однородности по переменным п, N должен иметь вид и26 (Р, T)/2N, где В — функция только от Р и Т. Таким образом, с точностью до членов второго порядка термодинамический потенциал слабого раствора имеет вид
Ф = ЛГр,„(Р, Т)+пТ\п^ + пЦ(Р, Г)+^В(Р, Т). (87,2)
Обобщение этого выражения на случай раствора нескольких веществ очевидно:
Ф = iv>„ +£ п,Т In ^ + £ п,Ъ + Ц ^ в,*,
i i I, k
(87,3)
где щ—числа молекул различных растворенных веществ.
Из (87,1) легко найти химические потенциалы для растворителя (р) и растворенного вещества (р') в растворе:
дФ ON
| Г |
п.
Mo—Тс,
(87,4)
.' = %.= Т1п± + Ъ = Т1пс + Ъ.
(87,5)
§ 88. Осмотическое давление
В этом и в следующих параграфах мы рассмотрим некоторые свойства растворов, причем по-прежнему будем считать раствор слабым и потому будем пользоваться формулами предыдущего параграфа.
Предположим, что два раствора одного и того же вещества в одном и том же растворителе, но обладающие различными концентрациями сх и с2, отделены друг от друга перегородкой, сквозь которую могут проникать молекулы растворителя, но не растворенного вещества (полупроницаемая перегородка). Давления с обеих сторон перегородки будут при этом различными (рассуждения в § 12 о равенстве давлений здесь неприменимы благодаря наличию полупроницаемой перегородки). Разность этих давлений носит название осмотического давления.
Условием равновесия между обоими растворами будет (кроме равенства их температур) равенство химических потенциалов растворителя в них. Химические потенциалы растворенного вещества при этом не должны быть одинаковы, так как вследствие полупроницаемости перегородки равновесие имеет место только по отношению к растворителю.
Обозначая давления в обоих растворах буквами Рг и Р2 и воспользовавшись выражением (87,4), получим условие равновесия в виде
|i.(Plt Т)-с1Г = р0(Яа, Т)-с2Т. (88,1)
Разность давлений Р2— Pt=AP (т. е. осмотическое давление) для слабых растворов относительно мала. Поэтому можно разложить р0(Р2> Т) в ряд по степеням АР и оставить только два первых члена:
р0(Р2, Т) = М/\, Т)+АРд$.
Подставляя это в (88,1), находим
bPd$ = (c,-cJT.
Но производная Зр0/ЗР есть молекулярный объем v чистого растворителя. Таким образом,
AP = (c2-Cl)|. (88,2)
В частности, если с одной стороны перегородки находится чистый растворитель (^ = 0, с2 = с), то осмотическое давление
сТ пТ
где п есть число молекул растворенного вещества в объеме V растворителя (ввиду слабости раствора V с большой точностью равно полному объему раствора). Формулу (88,3) называют формулой Вант-Гоффа. Следует обратить внимание на то, что она применима к слабым растворам независимо от конкретного рода веществ (как растворителя, так и растворенного вещества), а также на сходство этой формулы с уравнением состояния идеального газа. Вместо давления газа здесь стоит осмотическое давление, вместо объема газа — объем раствора и вместо количества частиц в газе—количество молекул растворенного вещества.
§ 89. Соприкосновение фаз растворителя
Рассмотрим равновесие двух соприкасающихся фаз растворителя, в каждой из которых растворено некоторое количество одного и того же вещества. Условиями равновесия являются (кроме равенства давлений и температур) равенства химических потенциалов растворителя и растворенного вещества в обеих фазах. Мы воспользуемся здесь первым из них и напишем его в виде
p.J(P, T)-ClT = p0I(P, Т)-СцТ, (89,1)
где С\ и Сц—концентрации, а р0 и р"—химические потенциалы обеих фаз чистого растворителя.
Надо заметить, что рассматриваемая нами теперь система, состоящая из двух компонент и имеющая две фазы, обладает двумя термодинамическими степенями свободы. Поэтому из четырех величин Р, Т, Ci, Сц только две можно выбрать произвольно; если мы выберем, например, Р или Т и одну из концентраций, то другая концентрация будет при этом определена.
Если бы обе фазы растворителя не содержали растворенного вещества, то условием их равновесия было бы
pi (Р0, Та) = р." (Ра, Тц) (89,2)
(температуру и давление обеих фаз мы при этом обозначили через Т0 и Р0)."
Таким образом, в то время как при равновесии фаз чистого растворителя зависимость между давлением и температурой определяется уравнением (89,2), после растворения в этих фазах какого-либо вещества та же зависимость определяется уравнением (89,1). Для слабых растворов обе эти кривые близки друг к другу.
Разложим теперь в равенстве (89,1) \il(P, Т) и р" (Р, Т) по степеням Р—Р0 = АР и Т — 70 = А7, где Р0 и Т9—давление и температура в некоторой точке на кривой равновесия фаз чистого растворителя, близкой к данной точке Р, Т на кривой равновесия фаз раствора. Оставляя в разложении только члены первого порядка относительно ДР и ДТ и принимая во внимание (89,2), получим из (89,1)
|? ДТ + ^ АР-схТ = дт + АР—спТ.
Но —фо/дТ и d\i0/dP не что иное, как энтропия s и объем v чистого растворителя (отнесенные к одной молекуле). Приписывая им также индекс, указывающий фазу, находим
- (s,-s„) AT + (о, -»„) ДР = (с,- с„) Т. (89,3)
Согласно формуле (81,5) имеем: (sn — si)T = q, где ^—теплота перехода растворителя из первой фазы во вторую. Поэтому (89,3) можно переписать также и в виде
f ДТ + (о,-г;11)АР = (с1-с„)Т. (89,4)
Разберем два частных случая этой формулы. Выберем сначала точку Р„, Т0 так, чтобы Р0 = Р. Тогда ДТ будет расстоянием по оси абсцисс между обеими кривыми при одной и той же ординате. Другими словами, ДТ будет представлять собой изменение температуры перехода между двумя фазами при растворении, т. е. разность между температурой Т этого перехода (при давлении Р), когда обе фазы являются растворами, и температурой Т0 перехода (при том же давлении) для чистого растворителя. Так как при этом ДР = 0, то из (89,4) получим
АТ = Г2(С1~С|1). (89,5)
Я
Если одна из фаз (скажем, первая) является чистым растворителем (с„ = 0, Cj = C), то
АТ = ^. (89,6)
Эта формула определяет, в частности, изменение температуры замерзания при растворении, если растворенное вещество нерастворимо в твердой фазе; двумя фазами являются при этом жидкий раствор и твердый растворитель, а ДТ есть разность между температурой вымерзания растворителя из раствора и температурой замерзания чистого растворителя. При замерзании тепло выделяется, т. е. q отрицательно. Поэтому и AT < О, т. е. если вымерзает чистый растворитель, то растворение понижает температуру замерзания.
Соотношение (89,6) определяет также изменение температуры кипения при растворении, если растворенное вещество не летуче: двумя фазами являются при этом жидкий раствор и пар растворителя. ДТ есть теперь разность температуры выкипания раства-
рителя из раствора и температуры кипения чистого растворителя. Поскольку при кипении теплота поглощается, то q > 0, а потому и ДТ > О, т. е. температура кипения при растворении повышается.
Все эти следствия из формулы (89,6) находятся в полном согласии с принципом Ле-Шателье. Пусть, например, жидкий раствор находится в равновесии с твердым растворителем. Если увеличить концентрацию раствора, то, согласно принципу Ле-Шателье, должна понизиться температура замерзания так, чтобы часть твердого растворителя перешла в раствор и концентрация понизилась. Система как бы противодействует выведению ее из состояния равновесия. Аналогично, если увеличить концентрацию Жидкого раствора, находящегося в равновесии с паром растворителя, то температура кипения должна повыситься так, чтобы часть пара сконденсировалась в раствор и концентрация понизилась.
Рассмотрим теперь другой частный случай формулы (89,4), выбрав точку Р0, Г0 так, чтобы Т = Т0, Тогда АР будет расстоянием между обеими кривыми при одной и той же абсциссе, т. е. разностью между давлением при равновесии двух фаз растворов и двух фаз чистого растворителя (при одной и той же температуре). Теперь ДТ = 0, и из (89,4) получаем
АР
_ Т(сх— сп)
(89,7)
Применим эту формулу к равновесию между жидкой и газообразной фазами. В этом случае объемом одной фазы (жидкой) можно пренебречь по сравнению с объемом другой, и (89,7) переходит в
АР = Г(СК~С|1), (89,8)
где v—молекулярный объем газообразной фазы (фаза I). Замечая, что Pv = T, и подставляя с той же точностью Р «Р0 (Р„ есть давление насыщенного пара над чистым растворителем), можно написать эту формулу в виде
Д/> = Р0(с,-с„).
(89,9)
Если газообразная фаза представляет собой пар чистого растворителя (с,= 0, сп = с), то (89,9) приобретает вид
АР
р- = —с,
(89,10)
где с—концентрация раствора. Эта формула определяет разность между давлением насыщенного пара растворителя над раствором (Р) и над чистым растворителем (Р0). Относительное понижение давления насыщенного пара при растворении равно концентрации раствора (закон Рауля)1).
§ 90. Равновесие по отношению к растворенному веществу
Далее рассмотрим систему, состоящую из двух соприкасающихся растворов одного и того же вещества в различных растворителях (например, в двух несмешивающихся жидкостях). Их концентрации обозначим буквами сг и с2.
Условием равновесия этой системы является равенство химических потенциалов растворенного вещества в обоих растворах. С помощью (87,5) это условие можно написать в виде
Пп^ + яМР, Т) = Т]пс, + $а(р, Т).
Функции и я|)2 для различных растворителей, конечно, различны. Отсюда находим
^ = exp(^i). (90,1)
Правая сторона этого равенства есть функция только от Р и Т. Таким образом, растворенное вещество распределяется между двумя растворителями так, чтобы отношение концентраций было (при заданных давлении и температуре) всегда одинаково, независимо от полного количества растворенного вещества и растворителей (закон распределения). Этот же закон относится, очевидно, и к растворению одного вещества в двух соприкасающихся фазах одного и того же растворителя.
Далее рассмотрим равновесие между газом (который будем считать - идеальным) и его раствором в некотором конденсированном растворителе. Условие равновесия, т. е. равенство химических потенциалов газа чистого и растворенного напишется (с помощью (42,6) и (87,5)) в виде
Т\пс + Ир(Р, Т) = Т)пР + %(Т),
откуда
с=Рехр(%=^у (90,2)
Функция ij)(P, Т) характеризует свойства жидкого (или твердого) раствора; однако при небольших давлениях свойства жидкости очень слабо зависят от давления. Поэтому и зависимость ■ф(Р, Т) от давления не играет роли, и можно считать, что
!) Напомним, что под с мы понимаем молекулярную концентрацию (отношение чисел молекул njN).
коэффициент при Р в (90,2) есть постоянная, не зависящая от давления:
с = Р- const. (90,3)
Таким образом, при растворении газа концентрация раствора (слабого) пропорциональна давлению газа (закон Генри)1).
Задачи
1. Найти изменение концентрации с высотой для раствора, находящегося
в поле тяжести.
Решение. Применим условие равновесия (85,3) во внешнем поле, причем напишем его для растворенного вещества: T\nc-\-ty(P, T)4-mgz = const, так как потенциальная энергия молекулы растворенного вещества в поле тяжести есть mgz (z— высота, m—масса молекулы). Продифференцируем это равенство по высоте, причем следует помнить, что температура постоянна (это—одно из условий равновесия):
Т dc,. дф dP п
Поскольку объем раствора равен ^^—N ^ (подставляем для Ф
выражение (87,1)), величину dty/dP можно назвать объемом v', приходящимся на одну молекулу растворенного вещества. Поэтому
Т dc.,,dP.
Чтобы найти зависимость Р от г, воспользуемся условием равновесия для растворителя 2)
где v = d]i0/dP — молекулярный объем, а М — масса молекулы растворителя. Подставляя dPjdz в предыдущее условие, находим
Т dc... v'.
Если раствор можно считать несжимаемым, т. е. v и v' постоянными, то отсюда находим формулу с=с0 ехр |—££^m_e_mjj (с0—концентрация раствора при z = 0), т. е. обычную барометрическую формулу, исправленную в соответствии с законом Архимеда.
| х) Подразумевается, что молекулы газа переходят в раствор в неизменном виде. Если при растворении молекулы распадаются (например, при растворении водорода Н2 в некоторых металлах), то зависимость концентрации от давления получается иной (см. задачу 3 к § 102). 2) Член с концентрацией (—Т dcjdz) в этом условии мал и может быть опущен (в условии для растворенного вещества он содержал с в знаменателе и потому не был мал). 3) Растворимость—концентрация насыщенного раствора. Предполагается, что эта концентрация все еще настолько мала, что применимы формулы теории слабых растворов. |
2. Найти связь между изменениями растворимостей двух веществ при их
одновременном растворении в одном растворителе3).
Решение. Взаимодействие между двумя растворенными веществами учитывается квадратичным (пропорциональным п-^п^) членом в термодинамическом потенциале (87,3). Химические потенциалы растворенных веществ
^ = wt=г 1п cl + ^+с^п + с^12
и аналогично для ц2 (концентрации c1 = n1/N, c2 = n2/N). Растворимости c0i и с02 каждого из веществ в отсутствие другого определяются условиями равновесия
|
|
|
|
|
Дата добавления: 2014-11-07; Просмотров: 413; Нарушение авторских прав?; Мы поможем в написании вашей работы!