КАТЕГОРИИ:
Архитектура-(3434)Астрономия-(809)Биология-(7483)Биотехнологии-(1457)Военное дело-(14632)Высокие технологии-(1363)География-(913)Геология-(1438)Государство-(451)Демография-(1065)Дом-(47672)Журналистика и СМИ-(912)Изобретательство-(14524)Иностранные языки-(4268)Информатика-(17799)Искусство-(1338)История-(13644)Компьютеры-(11121)Косметика-(55)Кулинария-(373)Культура-(8427)Лингвистика-(374)Литература-(1642)Маркетинг-(23702)Математика-(16968)Машиностроение-(1700)Медицина-(12668)Менеджмент-(24684)Механика-(15423)Науковедение-(506)Образование-(11852)Охрана труда-(3308)Педагогика-(5571)Полиграфия-(1312)Политика-(7869)Право-(5454)Приборостроение-(1369)Программирование-(2801)Производство-(97182)Промышленность-(8706)Психология-(18388)Религия-(3217)Связь-(10668)Сельское хозяйство-(299)Социология-(6455)Спорт-(42831)Строительство-(4793)Торговля-(5050)Транспорт-(2929)Туризм-(1568)Физика-(3942)Философия-(17015)Финансы-(26596)Химия-(22929)Экология-(12095)Экономика-(9961)Электроника-(8441)Электротехника-(4623)Энергетика-(12629)Юриспруденция-(1492)Ядерная техника-(1748)
Предполагаемое 4 страница
|
|
|
|
При выраженном лактат-ацидозе гипервентиляция с дыханием Куссмауля не редкость. Однако у некоторых больных с идентифицированным лакат-ацидозом присутствует гиповентиляция в той или иной степени, проявляющаяся относительной гиперкапнией. Дополнительное введение CO2 посредством бикарбоната будет способствовать усилению респираторного ацидоза, усугубляя текущее состояние больного.
Тем не менее, назначая ощелачивающую терапию растворами бикарбоната необходимо руководствоваться не только уровнем ацидемии по pH, но и уровнем HCO3- в крови больного. Выраженный ацидоз со снижением эндогенного бикарбоната менее 15 мэкв/л и нестабильной гемодинамикой может потребовать применения экзогенного бикарбоната. В таком случае допустимо введение половины рассчитанной дозы бикарбоната. Расчет производиться по формуле:
HCO3- = BE × 0,3 × mКГ
В одном литре 7,5% бикарбоната содержится 893 мэкв/л HCO3-, в 1 литре 8,4% бикарбоната – 1000 мэкв/л HCO3- (молярный раствор).
Карбикарб содержит значительно меньше CO2 и поэтому предпочтительнее бикарбоната (PaCO2 официнального раствора около 3 мм рт. ст.).
Применение методов искусственной респираторной поддержки необходимо проводить в режиме нормовентиляции под контролем PaCO2. Гипервентиляция увеличивает выведение CO2, вызывая развитие респираторного алкалоза. Алкалоз усиливает образование лактата, снижает активность ферментов аэробного гликолиза и вызывает вазоспазм, уменьшая тканевую перфузию.
Альтернативные методы лечения
Очень заманчивым и перспективным методом лечения лактат-ацидоза является применение дихлорацетата натрия. Дихлорацетат стимулирует пируватдегидрогеназу, ускоряя окисление пирувата до ацетил-КоА. В результате уровень лактата снижается, что подтверждено предварительными исследованиями[12]. Дополнительный эффект обусловлен положительным инотропным действием, что нивелирует супрессивное действие на миокард метаболического ацидоза. К сожалению, дихлорацетат относится к экзотическим лекарствам в отечественной медицине, находясь в тени из-за низкой восстребованности. Кроме того, он не запатентован, и ни одна из фармакологических компаний не имеет лицензии на его производство, что значительно снижает рыночную стоимость препарата и не приносит финансовой выгоды фирмам-производителям.
Пример.
Больной М. 64 года с острым инфарктом миокарда, кардиогенный шок, признаки левожелудочковой недостаточности и отека легких. Год назад перенес инфаркт миокарда заднедиафргамальной области. Постоянно принимает эналаприл, аспирин, последние несколько дней – фуросемид. АД – 80/60 на фоне прессорной поддержки Sol. Dobutamini 7 мкг/кг/мин.
Результаты КЩС-анализа:
pH = 7,29
PaCO2 = 48 мм рт. ст.
AB = 18,0 ммоль/л
SB = 16,0 ммоль/л
BE = - 10,2 ммоль/л
SpO2 = 88%
PaO2 = 67 мм рт. ст.
Результаты биохимического анализа плазмы:
Na+ = 135 ммоль/л
K+ = 5,5 ммоль/л
Cl- = 90 ммоль/л
Расчетный показатель:
AG = (135 + 5,5) – (18 + 90) = 32,5 ммоль/л
В данном случае по предварительному анализу у больного присутствует первичный метаболический ацидоз с сопутствующим респираторным ацидозом.
При использовании алгоритма оценки компенсации КЩС-расстройств (рис. 6 и 7) выявляется наличие у больного и дополнительного метаболического алкалоза.
Вычисление показателя «gap-gap» (разница разницы) подтверждает данное предположение. Отношение ΔAG/BE будет больше единицы:
ΔAG/BE ≈ 20/10 ≈ 2, то есть дефицит оснований значительно меньше от того, который должен был быть при таком количестве остаточных анионов (лактата). Такая ситуация может наблюдаться при сопутствующем метаболическом алкалозе (относительном избытке бикарбоната).
Метаболический алкалоз у данного больного развился вследствие употребления фуросемида. Фуросемид увеличивает поступление натрия в собирательные трубочки, что способствует интенсивному его обмену на калий и ион водорода, элиминирующиеся с мочой. Как правило, развивается гипокалиемия и метаболический алкалоз. Уровень калиемии 5,5 у больного свидетельствует о компенсаторном ацидоз-индуцируемом выходе калия из клеток.
Расчет анионной разницы говорит о наличии большого количества остаточных анионов, способствующих развитию метаболического ацидоза – в нашем случае лактата. Такое состояние обусловлено тканевой гипоперфузией вследствие резкого снижения сердечного выброса. Повреждение сердечной мышцы при инфаркте миокарда вызывает падение ударного объема левого желудочка. Сниженный сердечный выброс обусловливает развитие гипотонии, как следствие тканевую гипоперфузию и лактат-ацидоз.
В данном случае прямое измерение лактата в крови могло бы значительно облегчить установку диагноза. Однако в клинике, где лечился больной, измерение лактата не входило в перечень выполняемых лабораторией услуг. Следовательно, логично исключая другие причины увеличения анионной разницы и учитывая клинические данные, диагноз лактат-ацидоза является наиболее вероятным.
Больному была увеличена доза кардиотоника до 12 мкг/кг/мин, налажена подача увлажненного кислорода через носовой катетер. Измеренное насыщение гемоглобина кислородом при помощи пульсоксиметра составило 94%. Артериальное давление стабилизировалось до 105/70 мм рт. ст.
Начата внутривенная инфузия изотонического р-ра натрия хлорида.
Через 3 часа анализ КЩС крови и биохимического анализа крови фактически нормализовался, анионная разница составила 16 мэкв/л.
Однако, несмотря на проводимые лечебные мероприятия, через несколько часов больной скончался от фибрилляции желудочков с последующей асистолией. Реанимационные мероприятия в полном объеме, включая дефибрилляцию, успеха не принесли.
«Завтракайте плотнее, спартанцы, - ужинать нам сегодня придется на том свете». Царь Леонид перед битвой при Фермопилах
Кетоацидоз
| Метаболический ацидоз с увеличенной анионной разницей довольно часто возникает вследствие накопления кетоновых кислот – β-оксибутирата, ацетоацетата и ацетона. Кетоацидоз может развиться у людей болеющих сахарным диабетом, алкоголизмом либо при длительном голодании. |
Клиническая физиология и биохимия кетоновых тел
Кетоновые кислоты в обычных физиологических условиях вырабатываются в организме в ничтожно малых количествах (менее 0,1 ммоль/л). Являясь энергетически выгодным ресурсом, кетоновые кислоты могут представлять альтернативный источник клеточного топлива. В условиях сниженного питания происходит высвобождение свободных жирных кислот из адипоцитов, транспортировка их в печень и последующий метаболизм до кетоновых тел.
Кетоновые тела de novo синтезируются из ацетил-КоА, получаемого преимущественно при бета-окислении жирных кислот (см. рис.15). Дополнительными источниками кетонов являются пируват и кетогенные аминокислоты (чистые кетогенные – лизин и лейцин).
Реакции синтеза кетоновых кислот протекают в митохондриях гепатоцитов. Первые реакции, происходящие до образования ГМГ-КоА, могут проходить и в цитозоле, однако тогда ГМГ-КоА является промежуточным продуктом уже не в синтезе кетонов, а в синтезе холестерола. Цитозольный и митохондриальный ГМГ-КоА не смешиваются. Таким образом, ГМГ-КоА-синтетаза (обозначена цифрой 2 на рисунке 15) является обобщающим звеном как в синтезе холестерола, так и в синтезе кетоновых кислот.
β-оксибутират – это восстановленная форма ацетоацетата. В качестве кофактора этой реакции служит НАД-Н, в качестве фермента – β-оксибутиратсинтетаза. То есть, количество ацетоацетата, которое восстановится в β-оксибутират, напрямую зависит от соотношения НАД-Н/НАД (!).
Ацетон спонтанно образуется из ацетоацетата, количество ацетона прямо пропорционально количеству наработанного ацетоацетата.
Регуляция синтеза кетоновых кислот осуществляется при помощи инсулина и глюкагона. Прием пищи и выброс инсулина из β-клеток поджелудочной железы снижают образование кетоновых тел. Голодание усиливает липолиз, увеличивается уровень глюкагона. Уменьшение соотношения инсулин/глюкагон стимулирует кетогенез.
Избыток глюкагона облегчает транспорт ацил-КоА в митохондрии, что является определяющим этапом в окислении жирных кислот. Карнитин является своеобразным «транспортером» для ацил-КоА через митохондриальную мембрану, связываясь с ним под влиянием карнитилацилтрансферазы.
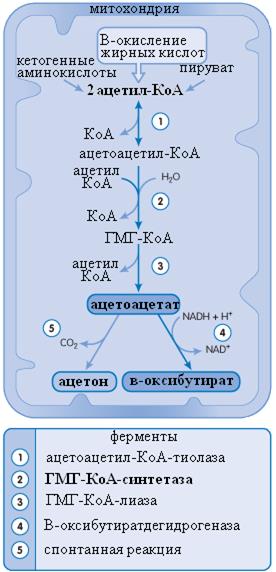
Рис. 15. Синтез кетоновых кислот. A. Clark, J. O’Neale Roach «CRASH COURSE: metabolism and nutrition» Elsevier Inc. Copyright © 2007 translated by KSS
Получившийся ацилкарнитин свободно проникает через внутреннюю митохондриальную мембрану, после чего ацил-КоА высвобождается и подвергается β-окислению, карнитин же возвращается к наружной
| Недостаточность инсулина |
| ▼ |
| Активация липолиза |
| ▼ |
| Повышение концентрации свободных жирных кислот в плазме |
| ▼ |
| Повышение концентрации свободных жирных кислот в печени |
| ▼ |
| УСКОРЕНИЕ КЕТОГЕНЕЗА |
| ▲ |
| Активация карнитилацилтрансферазы |
| ▲ |
| Повышение содержания карнитина в печени. Снижение содержания малонил-КоА |
| ▲ |
| Избыток глюкагона |
Рис. 16. Регуляция кетогенеза
митохондриальной мембране чтобы связаться со следующей молекулой ацил-КоА. Карнитилацилтрансфераза у сытого человека неактивна, в результате чего жирные кислоты не могут вступить в контакт с ферментами β-окисления. При голодании или декомпенсации диабета система транспорта жирных кислот активируется. Малонил-КоА является аллостерическим ингибитором активности карнитилацилтрансферазы. Глюкагон повышает содержание карнитина и снижает количество малонил-КоА, активируя тем самым карнитилацилтранферазу. В результате количество ацил-КоА, транспортированного в митохондрии, резко возрастает, стимулируя β-окисление жирных кислот. Продукт β-окисления – ацетил-КоА – метаболизируется по указанным выше путям, приводя в итоге к увеличению количества кетоновых кислот.
Ацетил-КоА в нормальных условиях метаболизируется циклом трикарбоновых кислот (цикл Кребса). При голодании либо декомпенсированном сахарном диабете оксалоацетат, необходимый для реакции с ацетил-КоА, чтобы формировать цитрат, используется в глюконеогенезе. Таким образом, организм пытается поддержать нормальный уровень гликемии при голодании или сахарном диабете («голод среди изобилия»). Но как следствие происходит накопление ацетил-КоА и неизбежный синтез кетоновых тел.
Итак, синтез кетоновых тел происходит в гепатоцитах, но использование их ограничивается только периферическими тканями. Например, для сердечной мышцы использование кетоновых кислот в качестве энергетического субстрата является более приоритетным по сравнению с использованием глюкозы. Активными потребителями кетоновых кислот являются также мышцы и головной мозг. Фактически утилизировать ацетоацетат и β-оксибутират может любая клетка, имеющая митохондрии. Например, эритроциты не имеют соответствующих клеточных структур, поэтому не способны метаболизировать кетоновые тела. Печень хоть и служит источником кетоновых кислот, но утилизировать их не способна, что обусловлено отсутствием в гепатоцитах фермента 3-кетоацил-КоА-трансферазы (см. рис. 17).
Энергетическая ценность кетоновых кислот
Ацетоацетат и β-оксибутират при
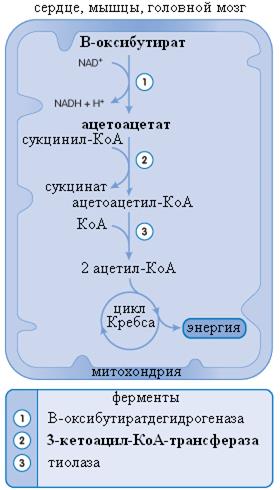
Рис. 17. Утилизация кетоновых тел
Жирным шрифтом выделен фермент, отсутствующий в печени (2). A. Clark, J. O’Neale Roach «CRASH COURSE: metabolism and nutrition» Elsevier Inc. Copyright © 2007 translated and changed by KSS
использовании их в качестве энергетического субстрата в клетках мышц, сердца или мозга подвергаются обратной трансформации в ацетил-КоА. Последовательность таких реакций отличается от реакций синтеза кетоновых кислот. β-оксибутират при помощи окисления НАД в НАД-Н превращается в ацетоацетат. Ацетоацетат при помощи 3-кетоацил-КоА-трансферазы переходит в ацетоацетил-КоА, затем образуются 2 молекулы ацетил-КоА и дальнейшее утилизация их в цикле трикарбоновых кислот (см. рис. 17).
Рассмотрим чистый выход энергии при утилизации одной молекулы β-оксибутирата. Как было сказано, одна молекула β-оксибутирата даёт две молекулы ацетил-КоА. Образованный в результате окисления кетоновых тел НАД-Н не идет в счет, так как равное количество НАД-Н расходуется при их синтезе. Так как для синтеза β-оксибутирата необходимо изначальное образование 2 молекул ацетил-КоА при окислении жирных кислот, то энергия, получаемая при этом учитывается и идет в общий энерговклад кетоновых тел – 8 молекул АТФ. Образовавшиеся все те же 2 молекулы ацетил-КоА при утилизации β-оксибутирата при последующем сгорании в цикле Кребса дают 20 молекул АТФ. Итого получаем 28 молекул АТФ, но так как для активации β-окисления жирных кислот необходимо затратить 2 АТФ, получаем конечный результат – 26 молекул АТФ. Для сравнения, утилизация одной молекулы глюкозы при аэробном гликолизе дает 36 АТФ. Как видно, энергетическая ценность кетоновых кислот довольно велика, что особенно важно при отсутствии поступления глюкозы в организм (длительное голодание). Количество образованных кетоновых кислот при голодании фактически равно необходимому для энергетической поддержки количеству, иными словами, приход равняется расходу.
Виды кетоацидоза
Существует три основные причины кетоацидоза – алкоголизм, голодание, декомпенсированный сахарный диабет.
Кетоацидоз вследствие голодания
Уменьшение поступления глюкозы в организм стимулирует активацию гликогенолиза и затем глюконеогенеза. После истощения запасов гликогена и субстратов глюконеогенеза запускается механизм липолиза. Липолиз на этом этапе является необходимым средством для поддержания энергетического обмена. Длинноцепочечные жирные кислоты поступают в гепатоциты, превращаясь в ацетил-КоА. Этому способствует обусловленная голодом и глюкагоном активация карнитилацилтрансферазы. Имеет место замедленная утилизация ацетил-КоА циклом Кребса вследствие дефицита расходующегося на глюконеогенез оксалоацетата. В совокупности вышеперечисленные причины приводят к увеличению синтеза кетоновых кислот.
Использование в активированном глюконеогенезе глюкогенных аминокислот (глицин, серин, аланин, глутамин) нарушает соотношение глюкогенные аминокислоты/кетогенные аминокислоты в пользу последних, также усиливая кетогенез. Кетоновые кислоты должны эффективно замещать нехватку энергии при дефиците углеводов. Однако вследствие дефицита оксалоацетата утилизация кетоновых кислот посредством цикла трикарбоновых кислот может быть затруднена. Тем не менее, баланс синтез/потребление кетонов практически полностью уравновешен, то есть ацетоацетат и β-оксибутират синтезируются в количествах, способных утилизироваться при данных условиях.
Возникающий при голодании кетоацидоз, как правило, несущественен, развивается через 3 дня голодания и редко достигает значительных величин.
Диагностика кетоацидоза вследствие голодания.
Диагностика кетоацидоза при длительном голодании не составляет трудностей. Анамнез, данные клинического осмотра и расчет анионной разницы играют решающую роль. Измерение концентрации кетоновых тел с помощью нитропруссидной реакции может быть затруднено (см. «алкогольный кетоацидоз, особенности диагностики»).
Лечение кетоацидоза вследствие голодания.
Лечение кетоацидоза, развившегося вследствие длительного голодания, заключается в обеспечении адекватного питания больного. Если позволяет состояние больного, возможно возобновление энтерального питания, стол №1 с добавлением содержащих глюкозу напитков – сладкий чай и пр. Объем кормления должен быть рациональным, дробными порциями, для адаптации кишечника к пищевой нагрузке. В случаях, когда провести энтеральное питание невозможно, начинается инфузия растворов глюкозы в дозировке 0,5 г/кг/час без добавления инсулина под контролем гликемии. Общий объем вводимой глюкозы рассчитывается из количества не менее 200 грамм в сутки. Возможно одновременное проведение энтерального питания и внутривенная инфузия глюкозосодержащих растворов. Смысл терапии кетоацидоза, обусловленного длительным голоданием – возобновить поставку глюкозы в организм, что задерживает продукцию кетоновых тел и предотвращает дальнейший катаболизм.
Алкоголик – это тот больной, который пьёт больше, чем его собственный врач.
A. L. Barash
Алкогольный кетоацидоз
Алкогольный кетоацидоз возникает вследствие определённых особенностей метаболизма этанола. Этиловый спирт метаболизируется в два основных этапа. Сперва под действием алкогольдегидрогеназы этанол превращается в ацетальдегид, последний при помощи альдегиддегидрогеназы метаболизируется в ацетат (см. рис. 18).
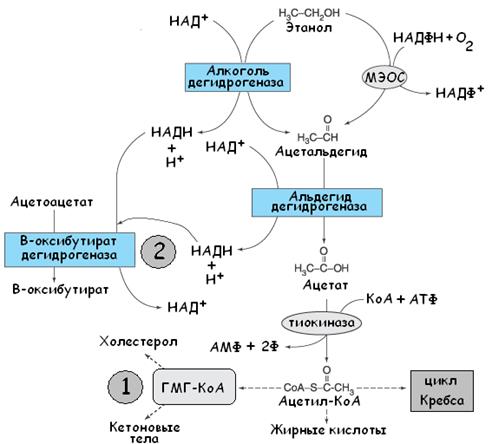
Рис. 18. Метаболизм этанола. Основные механизмы возникновения алкогольного кетоацидоза (обозначены цифрами). Created by KSS.
Ацетат, трансформируясь в ацетил-КоА, способствует синтезу кетоновых кислот (цифра 1 на рис. 18). Второй механизм активации кетогенеза – повышенная наработка НАД-Н. Первые два этапа метаболизма этилового спирта требуют окисления при помощи НАД+. Образующийся в большом количестве НАД-Н способствует переходу ацетоацетата в оксибутират (цифра 2 на рисунке 18). НАД-Н зависимым механизмом обладает и реакция перехода пирувата в лактат, что может привести к развитию лактат-ацидоза (см. лактат-ацидоз). Однако на практике чаще преобладает активация кетогенеза.
| При истощении запасов НАД+ реакция утилизация этанола переключается на путь микросомального окисления (см. рис. 18 – МЭОС – микросомальная этанолокисляющая система). При невысоких концентрациях этанола МЭОС практически не работает. Когда концентрация этанола превышает 100 мг%, влияние МЭОС возрастает вследствие истощения НАД+ - кофактора основного пути. В норме активность микросомальной этанолокисляющей системы составляет около 5% от активности всей ферментативной системы печени, метаболизирующей алкоголь. У хронических алкоголиков активность МЭОС приближается к 25%. Участие МЭОС в метаболизме осуществляется двумя путями. Первый из них заключается в непосредственном внедрении молекулярного кислорода в молекулу алкоголя с образованием соответствующего альдегида; второй связан с генерацией цитохромом P450 перекиси водорода, которая используется каталазой для окисления спирта. Метаболизм этанола подвергается кинетике нулевого порядка, то есть в определенную единицу времени печень способна переработать только определённое количество этанола. Это обусловлено именно НАД+-зависимым механизмом, а именно запасами НАД+ в гепатоцитах и его способностью к ресинтезу. Поскольку в реакции микросомального окисления этанола используется НАДФ-Н, довольно скоро наступает истощение и этого кофактора. Повышенное образование НАДФ и снижение НАДФ-Н препятствуют регенерации глутатиона. Глутатион является мощным антиоксидантом, «уборщиком» токсичных свободных радикалов, при снижении его количества печень оказывается незащищённой от оксидантного стресса. Так, ацетаминофен (парацетамол) при помощи системы p-450 в печени перерабатывается в гепатотоксичные метаболиты (N-ацетил-n-бензохимин), обладающие свойствами сильных электрофилов. Глутатион, связываясь с ацетаминофеном, метаболизируется до меркаптуровой кислоты, которая выводится с мочой. Поэтому риск повреждения печени при приеме препаратов содержащих ацетаминофен у страдающих алкоголизмом лиц несоизмеримо высок по сравнению со здоровыми людьми. Антидотом в таком случае является ацетилцистеин, способствующий восстановлению глутатиона в печени. |
Помимо вышеуказанных механизмов, зачастую у таких больных присутствует кетоацидоз вследствие голодания, поскольку болеющие хроническим алкоголизмом люди, как правило, испытывают алиментарную недостаточность (reduced nutrient intake) из-за асоциального образа жизни [1]. Не редкой находкой у таких больных является дегидратация, нарушающая экскрецию кетоновых тел с мочой.
Диагностика алкогольного кетоацидоза.
Алкогольный кетоацидоз (АКА) диагностируется в основном на основании клинических проявлений и наличии увеличенной анионной разницы необъяснимого генеза. Возникает обычно через 1-3 дня после чрезмерного потребления спиртных напитков. Клинически АКА проявляется тошнотой, рвотой и болью в животе. Уточнить диагноз помогает правильно собранный анамнез – наличие у больного длительного периода употребления спиртных напитков – запоя.
Характерны изменения электролитного состава плазмы, в частности, состояние «четыре гипо»: гипонатриемия, гипокалиемия, гипофосфатемия, гипомагниемия. Дегидратация наблюдается у всех больных тяжелым алкогольным кетоацидозом. Дефицит жидкости вызван длительной рвотой, потоотделением и полиурией. Полиурия обусловлена действием этанола, ингибирующего секрецию АДГ.
Отличительной чертой АКА от ДКА является нормальный или пониженный уровень гликемии. Возможно развитие смешанных нарушений КЩС, как правило, присоединение метаболического алкалоза вследствие частой рвоты и уменьшения количества внеклеточной жидкости (см. «метаболический алкалоз»).
Особенности диагностики.
Для лабораторной диагностики кетоновых тел в крови и моче используют нитропруссидную реакцию, выполняемую с помощью специальных тест-полосок или таблеток (Ацетест). Такая диагностика не является далекой от современных реалий большинства клиник страны и знать её суть и особенности обязан любой клиницист, так как понимание диагностики и клинической биохимии кетоновых кислот позволяет своевременно и правильно диагностировать нераспознанный кетоацидоз.
Диагностическая возможность нитропруссидного теста ограничивается концентрацией ацетоацетата около 3 ммоль/л. Если концентрация ацетоацетата будет ниже указанной цифры, тест будет отрицательный. Нитропруссидный тест позволяет выявить наличие только ацетоацетата. Если кетоацидоз обусловлен увеличением концентрации β-оксибутирата, тест будет отрицательным (см. рис. 19)
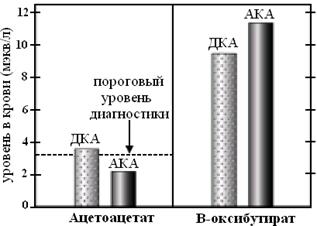
Рис. 19. Нитропруссидный тест в диагностике кетоацидоза. Прерывистой линией обозначена концентрация ацетоацетата, вызывающая положительную реакцию. ДКА – диабетический кетоацидоз, АКА – алкогольный кетоацидоз. P. L. Marino, ICU book, 2007
Повышенное образование НАД-Н у употребляющих алкоголь лиц способствует переходу ацетоацетата в β-оксибутират. Поэтому алкогольный кетоацидоз у них как правило обусловлен увеличением концентрации β-оксибутирата. Нитропруссидный тест у таких больных будет отрицательным, что, однако, не исключает наличия у них кетоацидоза. Соотношение β-оксибутират/ацетоацетат у больных с алкогольным кетоацидозом составляет 8:1, у больных с диабетическим кетоацидозом колеблется в пределах 3:1.
β-оксибутират превалирует в обоих случаях кетоацидоза, поэтому более точно следовало бы называть кетоацидоз β-оксибутират-ацидозом.
Определение ацетона в моче и в выдыхаемом воздухе может быть неинформативным. Ацетон образуется в ходе спонтанной реакции из ацетоацетата, а так как при алкогольном кетоацидозе β-оксибутират значительно преобладает над ацетоацетатом, то и количество ацетона не обязательно будет повышено. Необходимо помнить, что отрицательная нитропруссидная реакция и отсутствие ацетона в моче не могут полностью исключить алкогольный кетоацидоз. В таком случае помогает анамнез заболевания и расчет анионной разницы. Можно обойти проблему специфичности нитропруссидной реакции – достаточно добавить несколько капель перекиси водорода в пробу мочи. Это вызовет переход β-оксибутирата в ацетоацетат, который будет обнаружен нитропруссидным тестом.
Для кетоацидоза в целом и АКА в частности свойственно увеличение анионной разницы, однако эта зависимость не постоянна. Экскреция кетоновых тел почками повышает кислотность мочи. Снижение рН приводит к активации еще малоизученного Cl-/анионного обменника в проксимальных канальцах почек, который абсорбирует ионы хлора в обмен на выведенные анионы органических кислот. Повышение реабсорбции хлоридов поддерживает суммарный отрицательный заряд неизменным, но ограничивает рост анионной разницы.
Алкогольному кетоацидозу может сопутствовать лактат-ацидоз. Увеличение лактата происходит вследствие вышеописанной особенности метаболизма этанола, а также вследствие дегидратации и снижения тканевой перфузии. При уровне гликемии свыше 16,5 ммоль/л (300 мг/дл) можно говорить о сопутствующем диабетическом кетоацидозе (АКА у больных сахарным диабетом).
Кетоацидоз может вызывать увеличение уровня амилазы в крови, что может маскировать обострение хронического панкреатита у злоупотребляющих алкоголем больных.
Лечение алкогольного кетоацидоза
Лечение алкогольного кетоацидоза заключается в ликвидации трех основных патофизиологических синдромов:
· уменьшенный объем внеклеточной жидкости;
· истощенный запас гликогена;
· увеличенное соотношение НАДН/НАД+.
Конечная цель лечения - предотвращение образования кетонов и ускорение их выведения из организма. Применение буферирующих растворов не имеет смысла и в таких случаях не оправдано.
Введение растворов 5% глюкозы эффективно предотвращает выработку кетоновых тел, а инфузионная терапия изотоническими солевыми растворами ускоряет их экскрецию с мочой. Возможно применение сбалансированных растворов глюкозы и хлорида натрия (D5NS therapy – 5% dextrose in normal saline). Использование глюкозы у больных алкоголизмом может усугубить имеющийся дефицит тиамина и спровоцировать острое возникновение «бери-бери» или в особо тяжелых случаях – синдрома Вернике-Корсакова (Wernicke-Korsakoff syndrome). Поэтому при алкогольном кетоацидозе рекомендуется добавлять в растворы глюкозы витамин B1 в количестве 50-100 мг.
| Синдром Вернике-Корсакова по сути является двумя разными состояниями: энцефалопатией Вернике и Корсаковским синдромом, обусловленными одной причиной – дефицитом тиамина. При дефиците тиамина сперва развивается энцефалопатия Вернике, сопровождающаяся поражением нервных клеток и сосудов серого вещества, окружающих водопровод, III и IV желудочки головного мозга. Характерные признаки этого заболевания – офтальмоплегия, нистагм, атаксия и спутанность сознания. Энцефалопатия Вернике является обратимым состоянием, купирующимся после введения тиамина. В отсутствие лечения часто развивается хроническое состояние, при котором страдают обучаемость и память на фоне сохранности других психических функций. Такое состояние называется Корсаковским синдромом, характеризуется конфабуляциями и почти необратимо. Таким образом, Корсаковский синдром может являться спутником энцефалопатии Вернике и наблюдается в запущенных случаях дефицита тиамина. |
У больных алкогольным кетоацидозом зачастую запасы гликогена снижены, поэтому, во избежание эпизодов гипогликемии, инсулин не назначается. При необходимости производится коррекция электролитного состава плазмы. Алкогольный кетоацидоз обычно разрешается в течение 24 часов интенсивной терапии. В самом начале лечения возможно увеличения количества кетоновых тел в крови и моче, связанное со сдвигом реакций кетогенеза в сторону образования ацетоацетата.
Алкогольный кетоацидоз всегда необходимо дифференцировать от диабетического. Эти состояния могут встречаться одновременно, затрудняя своевременную диагностику.
Диабетический кетоацидоз
Диабетический кетоацидоз развивается у больных инсулинозависимым сахарным диабетом (diabetes mellitus, I тип) при декомпенсированном течении заболевания. У больных сахарным диабетом II типа кетоацидоз встречается крайне редко. Смертность при диабетическом кетоацидозе при своевременном лечении не превышает 2%. До того, как в 1922 году открыли инсулин, смертность составляла 100%. Среди взрослых людей, болеющих сахарным диабетом, ДКА является причиной смертности в 16% случаев. У детей, больных СД, такой показатель приближается к 70%.
Развитию кетоацидоза способствуют:
· нерациональная инсулинотерапия:
Ø недостаточная введенная доза;
Ø смена препарата без определения чувствительности к новому препарату;
|
|
|
|
|
Дата добавления: 2015-05-09; Просмотров: 2316; Нарушение авторских прав?; Мы поможем в написании вашей работы!